Volume 19, Number 7—July 2013
Letter
Spotted Fever Group Rickettsiae in Questing Ticks, Central Spain
Figure
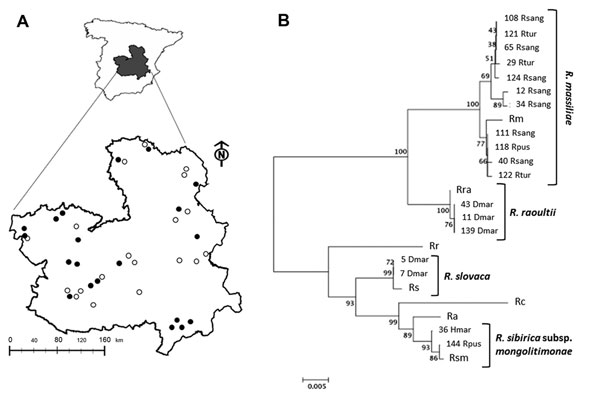
Figure. . Rickettsia species in questing ticks collected in central Spain. A) Study area with 20 collection sites where ticks were found (black dots) of the 39 sites surveyed (white and black dots). B) Multilocus sequence analysis of Rickettsia spp. The evolutionary history was inferred by using the neighbor-joining method of ompA-ompB concatenated sequences (total length = 1,189 nt). The optimal tree with the sum of branch length = 0.15227017 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic relationship. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. Codon positions included were 1st+2nd+3rd+noncoding. All ambiguous positions were removed for each sequence pair. Evolutionary analyses were conducted in MEGA5 (www.megasofware.net). The number of the Rickettsia spp. recognized in this study is shown next to the tick spp. Identified with them. Clusters of identified Rickettsia spp. are shown. Rc, Rickettsia conorii strain Malish 7; Ra, R. africae strain ESF-5; Rr, R. rickettsii strain Iowa; Rs, R. slovaca strain 13-B; Rm, R. massiliae strain MTU5; Rsm, R. sibirica subsp. mongolitimonae strain HA-91; R. raoultii isolate XG86; Rhsang, Rhipicephalus sanguineus; Rtur, Rh. turanicus; Rpus, Rh. pusillus; Dmar, Dermacenter marginatus; Hmar, Hyalomma marginatum. Scale bar indicates number of nucleotide changes per site.