Volume 2, Number 3—July 1996
Perspective
Molecular Approaches to the Identification of Unculturable Infectious Agents
Cite This Article
Citation for Media
Abstract
New molecular biologic techniques, particularly representational difference analysis, consensus sequence–based polymerase chain reaction, and complementary DNA library screening, have led to the identification of several previously unculturable infectious agents. New agents have been found in tissues from patients with Kaposi's sarcoma, non-A, non-B hepatitis, hantavirus pulmonary syndrome, bacillary angiomatosis, and Whipple's disease by using these techniques without direct culture. The new methods rely on identifying subgenomic fragments from the suspected agent. After a unique nucleic acid fragment belonging to an agent is isolated from diseased tissues, the fragment can be sequenced and used as a probe to identify additional infected tissues or obtain extended portions of the agent's genome. For agents that cannot be cultured by standard techniques, these approaches have proved invaluable for identification and characterization studies. Applying these techniques to other human diseases of suspected infectious etiology may rapidly elucidate novel candidate pathogens.
Identifying the causative agent of an infectious disease is the cornerstone for its eventual control. In recent years, a great deal of progress has been made in identifying new agents associated with both well-known and newly emerging infectious diseases. A number of syndromes exist, however, in which infectious etiology is likely, but the pathogen resists cultivation with standard microbiologic techniques. For emerging diseases, rapid identification and characterization of the responsible agent are crucial first steps for epidemic control.
The rapid identification of a hantavirus responsible for an outbreak of severe pulmonary distress syndrome in the southwestern United States (1, 2) demonstrated that applying molecular biologic approaches can accelerate the identification of an unknown agent. With extensive nucleic acid and protein databases readily available, isolating and sequencing genomic fragments from an unknown agent can provide important clues regarding its origin and biologic behavior. Once a new agent's phylogenetic relationship to other known organisms is established, appropriate culture conditions, serologic tests, and perhaps even therapeutic strategies can be rapidly developed.
Although they have revolutionized our ability to identify new pathogens, innovative molecular biologic techniques must be applied in conjunction with traditional epidemiologic procedures. Beral, Jaffe and colleagues, for example, showed that AIDS-related Kaposi's sarcoma (KS) is likely to be caused by a transmissible agent other than human immunodeficiency virus (HIV), before any likely causative agent was isolated (3, 4). These findings focused the attention of investigators on AIDS-KS, which resulted in the isolation of viral DNA from AIDS-KS lesions and the description of a new human herpesvirus (5, 6). Spurious associations between infectious agents and diseases are common, however, and only through careful epidemiologic studies can a causal link between an organism and a disease be established. Epidemiologic criteria for causality (7-9), superseding Koch's postulates, have been used for 30 years, and a critical phase in the process of new pathogen discovery involves the unambiguous establishment that an agent is central to the disease process.
The various molecular biologic approaches to agent identification differ in technical detail, but all rely on isolating nucleic acid fragments belonging to the agent's genome from diseased tissue. The formidable tasks of identifying and separating small unique nucleic acid fragments from human genomic material have been approached by various means, each with its own particular strengths and weaknesses. The appropriate technique depends on the type of infectious agent involved (e.g., bacterial or viral), whether the disease occurs in a normally sterile site, and whether it can be passaged through animals.
Once a fragment from the agent's genome is isolated and sequenced, standard genomic walking techniques are used to extend the known sequence, and computer homology searches can be used to identify the likely phylogenetic relationship of the agent to other known organisms. In this article, we highlight recent successful situations when molecular approaches were used to identify and characterize unknown agents of infectious diseases.
Figure
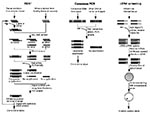
Figure. Schematic representation of different molecular approaches to the identification of unculturable infectious agents.
Representational difference analysis is one of the more robust methods of identifying new agents since it does not require prior knowledge of the agent's class (10). The technique is based on polymerase chain reaction (PCR) enrichment of DNA fragments present in diseased tissue but absent from healthy tissues of the same patient (Figure). Representational difference analysis is also an important tool for identifying polymorphic DNA sequences associated with noninfectious diseases (11).
Representational difference analysis depends first on digesting DNA from both healthy and diseased tissues by using a restriction enzyme and then on separately “simplifying” the resulting genomes to reduce their sequence complexity. This is done by ligating PCR primers to both sets of DNA and nonspecifically amplifying the mixtures. Since PCR most efficiently amplifies fragments of 150 to1500 bp, restriction fragments in this size range are enriched, and the fragments of the sequence represented outside this size range (90%) are reduced.
Unique strands of DNA from diseased tissue representing restriction fragments of an exogenous agent are isolated in a subtractive hybridization process coupled to PCR amplification. First, the priming sequences ligated on DNA restriction fragments of both normal and infected tissues are removed. New primer sequences are ligated only to the diseased tissue DNA fragments. These are then hybridized with an excess of the healthy tissue representation. Human DNA fragments common to both diseased and healthy tissues reanneal to each other and, since the healthy tissue fragments are in excess, any given human fragment derived from the diseased tissue will reanneal to a complementary strand from the healthy tissue representation. Thus, common human sequences found in both representations will only have one PCR priming sequence or none present on two complementary strands. However, DNA fragments from the infectious agent will not find complementary strands in the healthy tissue representation and will reanneal with each other. Only hybrids with both strands derived from the diseased tissue representation will have priming sites and be able to undergo subsequent exponential PCR amplification. Several rounds of representational difference analysis are performed, which successively enrich the mixture for unique DNA sequences present only in the diseased tissue representation.
KS-Associated Herpesvirus and KS
The power of representational difference analysis and the difficulties encountered in establishing the etiology of disease are illustrated by its application to KS (5), a vascular neoplasm that frequently occurrs in homosexual men with AIDS (3). Geographic clustering (12, 13) and association with specific sexual behavior (4, 14) suggest that the disease is caused by a sexually transmitted agent. Several agents have been investigated, including human cytomegalovirus (CMV), human papillomavirus, human herpesvirus 6 (HHV-6), and HIV (for review, see [15]), but no convincing etiologic link has been established.
Using representational difference analysis, Chang and colleagues isolated two unique DNA sequences (KS330Bam and KS631Bam) from a KS lesion in an AIDS patient (5). These DNA sequences are homologous to portions of minor capsid and tegument protein genes from EpsteinBarr virus (EBV) and herpesvirus saimiri (HVS), a New World monkey herpesvirus. Both EBV and HVS are gamma herpesviruses associated with neoplastic disorders in humans (EBV) (16) and nonhuman primates (HVS) (17). These results suggested that a new human herpesvirus, now called KS-associated herpesvirus (KSHV or HHV8), could be the KS agent.
The strong association between KS and KSHV was demonstrated by using Southern hybridization and PCR to amplify a 233-bp DNA fragment (KS330233) internal to KS330Bam from all 25 intact and amplifiable DNA samples from KS lesions that were examined (5). Ninety tissues from patients without AIDS or KS were examined, and none showed evidence of KSHV infection.
Although controversy remains over the role of KSHV in KS (18-20), epidemiologic and biologic studies strongly suggest that the agent is a causal factor in KS (21). KSHV has now been identified in 211 (94.2%) of 224 KS lesions examined by a number of groups around the world, and the virus is found in all forms of KS, AIDS-related and non-AIDS-related (22-29). Two independent studies have shown that KSHV is present in peripheral blood mononuclear cells of AIDS-KS patients before onset of KS (30, 31), indicating that the virus is unlikely to be a “passenger virus” in KS lesions. Further, the virus has been localized to KS tumor tissues by semiquantitative PCR (25) and by PCR in-situ hybridization (26). The virus has only been found in 8 (1.8%) of 449 solid tissues from control patients without KS (5, 22, 25, 27, 28, 32, 38) and thus appears to be specifically associated with KS. Although early reports suggested that the virus is present in skin tumors from transplant patients without KS (19), subsequent studies have not confirmed this finding (33, 34). One study has found evidence of viral DNA by nested PCR, but not unnested PCR, in semen from (23%) of healthy donors; these results suggest that the prevalence of infection in North America may be higher than that indicated by tissue or lymphocyte studies (35). This intriguing finding has not been reproduced by other groups (23, 36); however, it should be explored in large rigorous studies.
Lymphoproliferative disorders are common secondary malignancies in KS patients with and without AIDS (37). KSHV has also been found in a rare subset of AIDS-related, body cavity-based lymphomas, which are manifested as primary effusions (5, 38). In these AIDS-related lymphomas, tumor cells are coinfected with EBV (38, 39) but, unlike tumor cells in EBV-related Burkitt's lymphomas, these cells do not exhibit c-myc gene rearrangement. EBV-uninfected, KSHV-related body cavity-based lymphomas have also recently been identified (40, 43). Another lymphoproliferative syndrome associated with KS is Castleman's disease in both AIDS patients and HIV-seronegative patients. KSHV has been found in AIDS-related multicentric Castleman's disease lesions at a high copy number and less frequently in tissues from Castleman's disease patients without AIDS (41).
Identifying KSHV DNA sequences by representational difference analysis also led to the quick identification of an in vitro culture system for growing the virus. KS330Bam and KS631Bam probes were used to identify a B-cell line derived from a body cavity-based lymphoma that was stably infected with both KSHV and EBV (42). Extended sequencing and transmission studies have been performed with body cavity-base lymphoma cell lines, which clearly define KSHV as a new human herpesvirus of the genus Rhadinovirus (6). This has recently been confirmed by detecting herpesvirus particles in a body cavity-based lymphoma cell line by electron microscopy (43).
Discovery of a continuously infected cell line has allowed the first generation of serologic assays for KSHV antibodies to be developed (6). Second-generation immunoblotting assays, which appear to be both sensitive and specific for detecting KSHV antibodies, indicate that KS patients are infected with the virus months to years before the disease develops and that few North American blood donors are infected (S-J Gao, P.S. Moore, unpublished observation). Thus, identifying a virus associated with KS by using molecular approaches has rapidly led to new assays that use traditional serologic techniques for detecting infection.
HHV-6 and Multiple Sclerosis
The difficulties of using representational difference analysis for new pathogen identification are also illustrated by the search for the agent that causes multiple sclerosis (MS) (44). An infectious cause for MS has been proposed (45-47), and geographic and household clustering of cases consistent with an infectious process have been shown (45, 48-50). Challoner and colleagues used DNA from sclerotic plaques in brain tissues from MS patients and performed representational difference analysis against pooled DNA from peripheral blood leukocytes of healthy donors. Use of pooled lymphocyte DNA in representational difference analysis opens the possibility for examining rare banked tissues for which healthy control tissues are not available. A 341-bp representational difference analysis band was isolated from MS tissues with near sequence identity to a region of the major DNA-binding protein gene of HHV-6 (44). Detecting exogenous DNA in diseased tissue by this technique, however, does not exclude the possibility that a commensal agent may be identified that is not the cause of the disease being examined. HHV-6 is a neurotrophic virus present in brain tissues from healthy control patients (51), and more than (70%) of both MS patients and controls were positive for the representational difference analysis sequence by PCR (44). Although case-patients and controls cannot be differentiated by the presence or absence of these HHV-6 sequences, only MS tissues showed nuclear staining of oligodendrocytes surrounding plaques when monoclonal antibodies against HHV-6 viral proteins were used (44). There may be a subtle difference in tissue distribution for the virus in MS patients not found in controls, or the HHV-6 variant B group 2 infecting MS plaques may be particularly prone to generating the autoimmune response seen in this disease.
GB Hepatitis Viruses
Since representational difference analysis relies on PCR amplification, the technique is suited for detecting agents with a DNA-based genome; sequences can be amplified by generating a cDNA intermediate, but detecting RNA viruses is problematic since mRNA expression patterns differ between tissues and are likely to give spurious representational difference analysis bands. Simons and colleagues overcame this problem by passing an agent associated with non-A, non-B (NANB) hepatitis through primates and using cell-free extracts of primate plasma for representational difference analysis and discovered two new human hepatitis viruses (52, 53).
A novel form of NANB hepatitis was first identified in a physician who became ill with hepatitis, and the disease was transmitted to primates by injecting serum from patients into the animals (54). Extensive studies demonstrated that the virus, named the GB agent, was different from all known human hepatitis viruses (hepatitis A [HAV], hepatitis B [HBV], hepatitis C [HCV], and hepatitis E) (55-57). Total RNA from the samples was reverse-transcribed to obtain cDNA by using cell-free preinfection and acute-phase plasma from infected monkeys. After cDNA synthesis, representational difference analysis was performed, and seven cDNA clones were found to be specifically associated with this form of hepatitis. Sequence analysis and comparison with other known hepatitis viruses identified two unique flaviviruses, GBVA and GBVB, as the GB agents (52, 53, 58). A third virus, GBVC, was subsequently identified using the known GBVA, GBVB, and HVC consensus sequences (52b and see below). GBVA and GBVC are closely related phylogenetically. Cross-challenge experiments showed that GBVC probably originated in human hepatitis patients, whereas GBVA and B may be tamarin monkey viruses that were inadvertently passaged along with the human virus (53, 58).
In addition to having difficulty in identifying RNA viruses, representational difference analysis has several other limitations. Polymorphic human DNA can be amplified through this technique (5), and not all bands generated by it belong to the suspected agent. Representational difference analysis may produce DNA fragments from an agent whose genome has not been sequenced (e.g., most bacteria and fungi), and sequence homology searches may be unable to distinguish the agent's genomic DNA fragments from unsequenced human DNA. Only normally sterile site tissues are appropriate for this technique because normal flora that differs between tissue sites could result in spurious amplification. Finally, for viruses with small genomes, multiple restriction digests may be required to identify a unique restriction fragment of the appropriate size that can be efficiently amplified. Although the technique has been successfully used by several groups to identify polymorphic DNA, the procedure is complex and not uniformly reproducible.
Consensus sequence PCR relies on the use of highly conserved DNA sequences, such as ribosomal RNA (rRNA) gene sequences from known organisms, to amplify DNA from related organisms not yet discovered (59). This technique is simple and extremely successful in identifying new human pathogens. Subunit rRNA genes evolve in a relatively slow and uniform manner, which makes these sequences extremely useful for establishing phylogenetic relationships (59). By using conserved DNA sequences from bacterial 16S rRNA genes, at least two new bacteria associated with human diseases have been identified (60, 61), and PCR amplification of conserved hantaviral capsid DNA sequences resulted in the rapid identification of a new hantavirus associated with an outbreak of severe pulmonary disease (1).
Unlike using representational difference analysis, using consensus sequences to amplify DNA requires knowledge of the suspected agent's phylogenetic relationship to other organisms. The technique generally should be used on normally sterile site tissues if sequences from normal flora are also likely to be amplified. Although the lack of broadly amplifiable consensus primers limits use of this technique to prokaryotic and eukaryotic pathogens, consensus sequences are to likely exist among many of the classes of viruses that eventually could be used in screening panels of diseased tissues when a viral cause is suspected.
Bartonella: Bacillary Angiomatosis and Cat-Scratch Disease
Phylogenetic studies of bacteria have relied on comparisons of highly conserved rRNA gene sequences present in prokaryotes (62). rRNA genes are present in all living cells and contain regions of highly conserved sequences with intervening variable regions. Conserved sequences can be used to amplify and sequence the intervening variable regions by PCR. This technique was exploited by Relman and colleagues to identify the bacillus associated with bacillary angiomatosis (60).
Bacillary angiomatosis is an inflammatory vascular proliferative process that affects the skin, lymph nodes, and visceral organs of AIDS patients (63). Although bacilli can be identified by Warthin-Starry staining of lesions (64, 65), the suspected causal organism was resistant to cultivation by standard techniques. Consensus oligonucleotide primers complementary to the 16S rRNA genes of eubacteria were used to amplify 16S rRNA gene fragments directly from tissue samples (60). Phylogenetic analysis of the amplified DNA sequence showed that the organism belonged to the genus Rochalimaea (now renamed Bartonella) (60). Bartonella organisms were also cultured from bacillary angiomatosis lesions (66) and blood from bacteremic patients (67, 68), and serologic analyses have associated the organisms with cat-scratch disease (69). Current evidence suggests that bacillary angiomatosis in HIV-seropositive patients results from infection with either B. quitana or a newly described Bartonella species, B. henselae (68, 70), whereas cat-scratch disease is primarily caused by infection with B. henselae (71) (for review, see [72]).
Whipple's Disease
Consensus sequence PCR was also used to identify an organism associated with Whipple's disease, one of the most persistent mysteries in microbiology (61). Whipple's disease is a systemic illness, first described in 1907, characterized by arthralgias, diarrhea, abdominal pain, and weight loss (73). Rod-shaped bacilli were identified histologically in Whipple's disease lesions in the early 1960s (74), but the suspected bacteria were not culturable by standard techniques (73). The agent was found to be a gram-positive actinomycete (Tropheryma whippelii gen.nov.sp.nov.), unrelated to any characterized species when a bacterial 165 rRNA sequence was amplified and sequenced directly from infected tissue (61, 75).
Hantavirus Pulmonary Syndrome
Consensus PCR primers have been successfully used to identify and classify bacteria, and similar techniques can be used to diagnose new viral agents. In May 1993, an outbreak of unexplained acute respiratory illness with a high death rate occurred in the southwestern United States (76). In the initial phases of the investigation, the cause of the syndrome was not clearly known to be infectious. Serologic tests quickly detected crossreactive antibodies to known hantaviral antigens in the serum of patients; these results suggested that a previously unrecognized hantavirus was the cause of the disease (77). PCR primers, based on consensus sequences within the G2 protein coding region of the M segment of the genomes of known hantaviruses, were designed and used in nested reverse-transcription PCR to amplify a short segment of the viral genome from diseased tissues (1).
Sequence analysis of the PCR products showed that the amplicon differed from the other known hantaviral sequences by least 30%, and phylogenetic analysis demonstrated that the new virus is most closely related to Prospect Hill hantavirus (1), a zoonotic hantavirus endemic in North America (78). Hantavirus antigens have been detected in endothelial cells from patients (1, 79), and virus particles have been identified in infected pulmonary endothelial cells and macrophages (80). Deer mice (Peromyscus maniculatus) are the primary host for the virus (1), and the virus has been passaged through laboratory-bred deer mice and cultured in Vero E6 cells (2). This newly recognized virus was originally named Muerto Canyon virus (2) but has since been renamed Sin Nombre virus in light of nomenclatural disputes regarding the appropriateness of a descriptive name.
Identification of HCV and HGV
A third major approach to new organism identification relies on screening cDNA libraries made from diseased tissues by using hyperimmune serum from specimens. This method was successfully used to identify the cause of most cases of NANB hepatitis, HCV. When serologic tests for HAV and HBV became available, it became clear that most cases of transfusion-associated hepatitis in the United States were not caused by either virus (81, 82). Conventional techniques failed to identify the agent responsible for most cases of NANB hepatitis (83), despite evidence that the disease was caused by a bloodborne, small, enveloped virus readily transmissible to chimpanzees (84, 85).
Since the virus was presumed to be an RNA virus, Choo and colleagues made a cDNA expression library from RNA isolated from an infected chimpanzee's plasma (86). While the GBVA, B, C were identified by direct detection of viral cDNA through representational difference analysis, HCV cDNA was identified by immunologic detection of cDNA that was encoding viral protein. Viral antigens expressed from the cDNA library were identified by immunoscreening with convalescent-phase human sera. A cDNA clone was isolated encoding an antigen that could be used to screen convalescent-phase sera from patients with NANB hepatitis. Identification of the clone rapidly led to the production of a recombinant antigen used for serologic screening to detect specific antibodies in infected chimpanzees and patients with hepatitis after transfusion. Extended sequence analysis demonstrated that the agent, now known as HCV, is closely related to the family Flaviviridae and is the major cause of NANB hepatitis throughout the world (86, 87). Recently, the same approach was successfully used to identify HGV, which has been found to be almost identical to GBVC, the human hepatitis virus identified by representational difference analysis (88). The potential role of HGV and GBVC in human disease still remains uncertain (88b).
Using convalescent-phase sera to screen cDNA libraries from diseased tissues is a novel method for pathogen identification. It is a potentially useful technique for diseases in which well-defined convalescent-phase sera are available, and it requires tissues with a high titers of the agent. On the other hand, constructing and screening DNA libraries are laborious, and crossreactive antigens are likely to be detected, especially for diseases in which autoantibodies are common.
Nucleic acid database information is rapidly expanding for all classes of organisms, and a significant fraction of the human genome has already been sequenced. Even small laboratories can exploit new and relatively inexpensive molecular biologic technologies to search for new pathogens. Once a small, unique nucleic acid fragment from a pathogen has been identified, nucleic acid detection and serologic assays can often be rapidly developed to establish an etiologic link with disease.
New pathogens are likely to be identified by some of these molecular biologic approaches. A number of diseases have long been suspected to have an infectious cause and are appropriate candidates for these techniques. The eventual identification of infectious etiologic agents of diseases such as sarcoidosis (89), Kawasaki's disease (90), and type I diabetes mellitus is likely. New techniques, such as arbitrarily primed PCR and phage display libraries (91), have not been used for pathogen discovery but show promise for expanding the repertoire of techniques available for identifying unculturable agents. Three years passed between the initial descriptions of AIDS and the identification of HIV (92). Use of molecular biologic approaches could lead to rapid identification and control of the next pandemic caused by a newly emergent infectious disease.
Dr. Gao received his Ph.D. in virology from the University of Bordeaux, Bordeaux, France. He is a research scientist in the School of Public Health, Columbia University, New York, where his work focuses on the serology and seroepidemiology of Kaposi's sarcomaassociated herpesvirus (KSHV). He also works on the induction of lytic phase replication of KSHV as well as genes that may be involved in the latent phase replication of the virus.
Dr. Moore is an associate professor of clinical public health at Columbia University. His current research is focused on delineating the relationship between Kaposi's sarcoma and KSHV (HHV8) and understanding the tumorigenic potential of this virus. Along with Dr. Yuan Chang, his laboratory codirector and wife, he is working on both epidemiologic and basic virologic studies of KSHV and other new potential pathogens.
Acknowledgment
We thank Drs. David Relman, Stanford University, and Yuan Chang, Columbia University, for critical advice regarding new pathogen identification and comments on the manuscript, and Francis Zappa for help in preparing the manuscript.
References
- Nichol ST, Spiropoulou CF, Morzunov S, . Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science. 1993;262:914–7. DOIPubMedGoogle Scholar
- Elliott LH, Ksiazek TG, Rollin PE, Isolation of the causative agent of hantavirus pulmonary syndrome. Am J Trop Med Hyg. 1994;51:102–8.PubMedGoogle Scholar
- Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi's sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123–8. DOIPubMedGoogle Scholar
- Beral V, Bull D, Darby S, Risk of Kaposi's sarcoma and sexual practices associated with faecal contact in homosexual or bisexual men with AIDS. Lancet. 1992;339:632–5. DOIPubMedGoogle Scholar
- Chang Y, Cesarman E, Pessin MS, . Identification of herpesviruslike DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;265:1865–9.
- Moore PS, Gao SJ, Dominguez G, . Primary characterization of a herpesviruslike agent associated with Kaposi's sarcoma. J Virol. 1996;70:549–58.PubMedGoogle Scholar
- Hill AB. Environment and disease: association or causation? Proc R Soc Med. 1965;58:295–300.PubMedGoogle Scholar
- Evans AS. Causation and disease: a chronological journey. Am J Epidemiol. 1978;108:249–58.PubMedGoogle Scholar
- Fredricks DN, Relman DA. Sequence-based identification of microbial pathogens: a reconsideration of Koch's postulates. Clin Microbiol Rev. 1996;9:18–33.PubMedGoogle Scholar
- Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–51. DOIPubMedGoogle Scholar
- Lisitsyn NA, Lisitsina NM, Dalbagni G, . Comparative genomic analysis of tumors: detection of DNA losses and amplification. Proc Natl Acad Sci U S A. 1995;92:151–5. DOIPubMedGoogle Scholar
- Archibald CP, Schechter MT, Craib KJ, Risk factors for Kaposi's sarcoma in the Vancouver Lymphadenopathy-AIDS Study. J Acquir Immune Defic Syndr. 1990;3(suppl 1):S18–23.PubMedGoogle Scholar
- Beral V, Bull D, Jaffe H, Is risk of Kaposi's sarcoma in AIDS patients in Britain increased if sexual partners came from United States or Africa? BMJ. 1991;302:624–5. DOIPubMedGoogle Scholar
- Archibald CP, Schechter MT, Le TN, Craib KJP, Montaner JSG, O'Shaughnessy MV. Evidence for a sexually transmitted cofactor for AIDS-related Kaposi's sarcoma in a cohort of homosexual men. Epidemiology. 1992;3:203–9. DOIPubMedGoogle Scholar
- Peterman TA, Jaffe HW, Beral V. Epidemiologic clues to the etiology of Kaposi's sarcoma. AIDS. 1993;7:605–11. DOIPubMedGoogle Scholar
- Harrington DS, Weisenburger DD, Purtilo DT. Epstein-Barr virus-associated lymphoproliferative lesions. Clin Lab Med. 1988;8:97–118.PubMedGoogle Scholar
- Rangan SR, Martin LN, Enright FM, Abee CR. Herpesvirus saimiri-induced lymphoproliferative disease in howler monkeys. J Natl Cancer Inst. 1977;59:165–71.PubMedGoogle Scholar
- Roizman B. New viral footprints in Kaposi's sarcoma. N Engl J Med. 1995;332:1227–8. DOIPubMedGoogle Scholar
- Rady PL, Yen A, Rollefson JL, . Herpesvirus-like DNA sequences in non-Kaposi's sarcoma skin lesions of transplant patients. Lancet. 1995;345:1339–40. DOIPubMedGoogle Scholar
- Levy JA. A new human herpesvirus: KSHV or HHV8? Lancet. 1995;346:786.
- Strathdee SA, Veugelers PJ, Moore PS. The epidemiology of HIV-associated Kaposi's sarcoma: the unraveling mystery. AIDS. 1995;10(suppl):S51–7.
- Moore PS, Chang Y. Detection of herpesviruslike DNA sequences in Kaposi's sarcoma lesions from persons with and without HIV infection. N Engl J Med. 1995;332:1181–5. DOIPubMedGoogle Scholar
- Ambroziak JA, Blackbourn DJ, Herndier BG, . Herpeslike sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science. 1995;268:582–3. DOIPubMedGoogle Scholar
- Collandre H, Ferris S, Grau O, Montagnier L, Blanchard A. Kaposi's sarcoma and new herpesvirus. Lancet. 1995;345:1043. DOIPubMedGoogle Scholar
- Dupin N, Grandadam M, Calvez V, Herpesviruslike DNA in patients with Mediterranean Kaposi's sarcoma. Lancet. 1995;345:761–2. DOIPubMedGoogle Scholar
- Boshoff C, Schulz TF, Kennedy MM, . Kaposi's sarcoma-associated herpesvirus (KSHV) infects endothelial and spindle cells. Nat Med. 1995;1:1274–8. DOIPubMedGoogle Scholar
- Chang Y, Ziegler JL, Wabinga H, Kaposi's sarcoma-associated herpesvirus DNA sequences are present in African endemic and AIDS-associated Kaposi's sarcoma. Arch Intern Med. 1996;156:202–4. DOIPubMedGoogle Scholar
- Schalling M, Ekman M, Kaaya EE, Linde A, Bieberfeld P. A role for a new herpesvirus (KSHV) in different forms of Kaposi's sarcoma. Nat Med. 1995;1:707–8. DOIPubMedGoogle Scholar
- Su I-J, Hsu Y-S, Chang Y-C, Wang IW. Herpesviruslike DNA sequence in Kaposi's sarcoma from AIDS and non-AIDS patients in Taiwan. Lancet. 1995;345:722–3. DOIPubMedGoogle Scholar
- Whitby D, Howard MR, Tenant-Flowers M, Detection of Kaposi's sarcoma-associated herpesvirus (KSHV) in peripheral blood of HIV-infected individuals predicts progression to Kaposi's sarcoma. Lancet. 1995;364:799–802. DOIGoogle Scholar
- Moore PS, Kingsley L, Holmberg SD, KSHV infection prior to onset of Kaposi's sarcoma. AIDS. 1996;10:175–80. DOIPubMedGoogle Scholar
- Boshoff C, Whitby D, Hatziionnou T, . Kaposi's sarcoma-associated herpesvirus in HIVnegative Kaposi's sarcoma. Lancet. 1995;345:1043–4.
- Adams V, Kempf W, Schmid M, Müller B, Briner J, Burg G. Absence of herpesvirus-like DNA sequences in skin cancers of non-immunosuppressed patients. Lancet. 1995;346:1715.
- Boshoff C, Talbot S, Kennedy M, New herpesvirus and immunosuppressed skin cancers. Lancet. 1996;347:338–9. DOIPubMedGoogle Scholar
- Lin JC, Lin SC, Mar EC, Is Kaposi's sarcoma-associated herpesvirus detectable in semen of HIV-infected homosexual men? Lancet. 1995;346:1601–2. DOIPubMedGoogle Scholar
- Gupta P, Mandaleshwar DS, Rinaldo C, Detection of Kaposi's sarcoma herpesvirus DNA in semen of homosexual men with Kaposi's sarcoma. AIDS. In press.
- Biggar RJ, Curtis RE, Cote TR, Rabkin CS, Melbye M. Risk of other cancers following Kaposi's sarcoma: relation to acquired immunodeficiency syndrome. Am J Epidemiol. 1994;139:362–8.PubMedGoogle Scholar
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesviruslike DNA sequences are present in AIDS-related body cavity based lymphomas. N Engl J Med. 1995;332:1186–91. DOIPubMedGoogle Scholar
- Knowles DM, Inghirami G, Ubriaco A. DallaFavera R. Molecular genetic analysis of three AIDS-associated neoplasms of uncertain lineage demonstrates their B-cell derivation and the possible pathogenetic role of Epstein-Barr virus. Blood. 1989;73:792–9.PubMedGoogle Scholar
- Nador RG, Cesarman E, Knowles DM, Said JN. Herpeslike DNA sequences in a body cavity-based lymphoma in an HIV-negative patient. N Engl J Med. 1995;333:943. DOIPubMedGoogle Scholar
- Soulier J, Grollet L, Oskenhendler E, . Kaposi's sarcoma-associated herpesviruslike DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276–80.PubMedGoogle Scholar
- Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. In vitro establishment and characterization of two AIDS-related lymphoma cell lines containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995;86:2708–14.PubMedGoogle Scholar
- Renne R, Zhong W, Herndier B, . Lytic growth of Kaposi's sarcoma-asociated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2:342–6. DOIPubMedGoogle Scholar
- Challoner PB, Smith KT, D . PJ, . Plaque-associated expression of human herpesvirus 6 in multiple sclerossis. Proc Natl Acad Sci U S A. 1995;92:7440–4. DOIPubMedGoogle Scholar
- Kurtzke JF. Some epidemiologic features compatible with an infectious origin for multiple sclerosis. Int Arch Allergy Appl Immunol. 1969;36:59–81.PubMedGoogle Scholar
- Cook SD, Dowling PC. Multiple sclerosis and viruses: an overview. Neurology. 1980;30:80–91.PubMedGoogle Scholar
- Whitaker JN, Kingsbury DW. Viruses and the pathogenesis of multiple sclerosis. Trends Neurosci. 1984;7:57–60. DOIGoogle Scholar
- Kurtzke JF. Familial incidence and geography in multiple sclerosis. Acta Neurol Scand. 1965;41:127–39. DOIPubMedGoogle Scholar
- Kurtzke JF. On the fine structure of the distribution of multiple sclerosis. Acta Neurol Scand. 1967;43:257–82. DOIPubMedGoogle Scholar
- Luppi M, Barozzi P, Maiorana A, Marasca R, Torelli G. Human herpesvirus 6 infection in normal human brain tissue. J Infect Dis. 1994;169:943–4.PubMedGoogle Scholar
- Simons JN, Pilot-Matias TJ, Leary TP, . Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc Natl Acad Sci U S A. 1995;92:3401–5. DOIPubMedGoogle Scholar
- Simons JN, Leary TP, Dawson GJ, . Isolation of novel virus-like sequences associated with human hepatitis. Nat Med. 1995;1:564–9. DOIPubMedGoogle Scholar
- Schlauder GG, Dawson GJ, Simons JN, Molecular and serologic analysis in the transmission of GB hepatitis agents. J Med Virol. 1995;46:81–90. DOIPubMedGoogle Scholar
- Deinhardt F, Holmes AW, Wolfe L, Junge U. Transmission of viral hepatitis to nonhuman primates. Vox Sang. 1970;19:261–9. DOIPubMedGoogle Scholar
- Tabor E, Peterson DA, April M, Seeff LB, Gerety RJ. Transmission of human non-A, non-B hepatitis to chimpanzees following failure to transmit GB agent hepatitis. J Med Virol. 1980;5:103–8. DOIPubMedGoogle Scholar
- Feinstone SM, Alter HJ, Dienes HP, Non-A, non-B hepatitis in chimpanzees and marmosets. J Infect Dis. 1981;144:588–98.PubMedGoogle Scholar
- Karayiannis P, Petrovic LM, Fry M, Studies of GB hepatitis agent in tamarins. Hepatology. 1989;9:186–92. DOIPubMedGoogle Scholar
- Relman DA. The identification of uncultured microbial pathogens. J Infect Dis. 1993;168:1–8.PubMedGoogle Scholar
- Relman DA, Loutit JS, Schmidt TM, Falkow S, Tompkins LS. The agent of bacillary angiomatosis: an approach to the identification of uncultured pathogens. N Engl J Med. 1990;323:173–80.PubMedGoogle Scholar
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple's disease. N Engl J Med. 1992;327:293–301.PubMedGoogle Scholar
- Lane DJ, Stahl DA, Olsen GJ, Heller DJ, Pace NR. Phylogenetic analysis of the genera Thiobacillus and Thiomicrospira by 5S rRNA sequences. J Bacteriol. 1985;163:75–81.PubMedGoogle Scholar
- Stoler MH, Bonfiglio TA, Steigbigel RT, Pereira M. An atypical subcutaneous infection associated with acquired immune deficiency syndrome. Am J Clin Pathol. 1983;80:714–8.PubMedGoogle Scholar
- Angritt P, Tuur SM, Macher AM, Epithelioid angiomatosis in HIV infection: neoplasm or cat-scratch disease? Lancet. 1988;1:996. DOIPubMedGoogle Scholar
- LeBoit PE, Berger TG, Egbert BM, Epithelioid haemangioma-like vascular proliferation in AIDS: manifestation of cat-scratch disease bacillus infection? Lancet. 1988;1:960–3. DOIPubMedGoogle Scholar
- Koehler JE, Quinn FD, Berger TG, LeBoit PE, Tappero JW. Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis. N Engl J Med. 1992;327:1625–31.PubMedGoogle Scholar
- Slater LN, Welch DF, Hensel D, Coody DW. A newly recognized fastidious gram-negative pathogen as a cause of fever and bacteremia. N Engl J Med. 1990;323:1587–93.PubMedGoogle Scholar
- Regnery RL, Anderson BE, Clarridge JE, RodriguezBarradas MC, Jones DC, Carr JH. Characterization of a novel Rochalimaea species, R. henselae sp. nov., isolated from blood of a febrile, human immunodeficiency virus-positive patient. J Clin Microbiol. 1992;30:265–74.PubMedGoogle Scholar
- Regnery RL, Olson JG, Perkins BA, Bibb W. Serological response to “Rochalimaea henselae” antigen in suspected catscratch disease. Lancet. 1992;339:1443–5. DOIPubMedGoogle Scholar
- Welch DF, Pickett DA, Slater LN, Steigerwalt AG, Brenner DJ. Rochalimaea henselae sp. nov., a cause of septicemia, bacillary angiomatosis, and parenchymal bacillary peliosis. J Clin Microbiol. 1992;30:275–80.PubMedGoogle Scholar
- Koehler JE, Glaser CA, Tappero JW. Rochalimaea henselae infections: a new zoonosis with the domestic cat as reservoir. JAMA. 1994;271:531–5. DOIPubMedGoogle Scholar
- Regnery RL, Tappero J. Unravelling mysteries associated with cat-scratch disease, bacillary angiomatosis, and related syndromes. EID. 1995;1:16–21. DOIGoogle Scholar
- Dobbins WO III. Whipple's disease. Springfield, IL: Charles C Thomas, 1987.
- Chears WCJ, Ashworth CT. Electron microscopic study of the intestinal mucosa in Whipple's disease: demonstration of encapsulated bacilliform bodies in the lesion. Gastroenterology. 1961;41:129–38.PubMedGoogle Scholar
- Wilson KH, Blitchington R, Frothingham R, Wilson JA. Phylogeny of the Whipple's disease-associated bacterium. Lancet. 1991;338:474–5. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Outbreak of acute illness—southwestern United States. MMWR. 1993;42:421–4.PubMedGoogle Scholar
- Centers for Disease Control and Prevention. Update: outbreak of hantavirus infection—southwestern United States. MMWR. 1993;42:441–3.PubMedGoogle Scholar
- Leduc JW. Epidemiology of Hantaan and related viruses. Lab Anim Sci. 1987;37:413–8.PubMedGoogle Scholar
- Ksiazek TG, Peters CJ, Rollin PE, Identification of a new North American hantavirus that causes acute pulmonary insufficiency. Am J Trop Med Hyg. 1995;52:117–23.PubMedGoogle Scholar
- Zaki SR, Greer PW, Coffield LM, Hantavirus pulmonary syndrome. Pathogenesis of an emergng infectious disease. Am J Pathol. 1995;146:552–79.PubMedGoogle Scholar
- Feinstone SM, Kapikian AZ, Purcell RH, Alter HJ, Holland PV. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N Engl J Med. 1975;292:767–70.PubMedGoogle Scholar
- Knodell RG, Conrad ME, Dienstag JL, Bell CJ. Etiological spectrum of post-transfusion hepatitis. Gastroenterology. 1975;69:1278–85.PubMedGoogle Scholar
- Shih JW, Mur JI, Alter HJ. Non-A, non-B hepatitis: advances and unfulfilled expectations of the first decade. Prog Liver Dis. 1986;8:433–52.PubMedGoogle Scholar
- Bradley DW, McCaustland KA, Cook EH, Schable CA, Ebert JW, Maynard JE. Posttransfusion non-A, non-B hepatitis in chimpanzees: physicochemical evidence that the tubule-forming agent is a small, enveloped virus. Gastroenterology. 1985;88:773–9.PubMedGoogle Scholar
- He LF, Alling D, Popkin T, Shapiro M, Alter HJ, Purcell RH. Determining the size of non-A, non-B hepatitis virus by filtration. J Infect Dis. 1987;156:636–40.PubMedGoogle Scholar
- Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–62. DOIPubMedGoogle Scholar
- Kuo G, Choo QL, Alter HJ, . An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–4. DOIPubMedGoogle Scholar
- Linnen J, Wages Jr J, Zhang-Keck ZY, . Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science. 1996;271:505–8. DOIPubMedGoogle Scholar
- Alter HJ. The cloning and clinical implications of HGV and HGBV-C. N Engl J Med. 1996;334:1536–7. DOIPubMedGoogle Scholar
- Graham DY, Markesich DC, Kalter DC, Moss MT, Hermon-Taylor J, El-Zaatari FA. Mycobacterial aetiology of sarcoidosis. Lancet. 1992;340:52–3.
- Rowley AH, Wolinsky SM, Relman DA, . Search for highly conserved viral and bacterial nucleic acid sequences corresponding to an etiologic agent of Kawasaki disease. Pediatr Res. 1994;36:567–71.PubMedGoogle Scholar
- Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–90. DOIPubMedGoogle Scholar
- Barre-Sinoussi F, Chermann JC, Rey F, . Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 1983;220:868–71. DOIPubMedGoogle Scholar
Figure
Cite This ArticleTable of Contents – Volume 2, Number 3—July 1996
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Shou-Jiang Gao, Ph.D., School of Public Health, Columbia University, P&S 14-442, 630 West-168th St., New York, NY 10032; fax: 212-305-4548
Top