Volume 20, Number 1—January 2014
Dispatch
Dobrava-Belgrade Virus in Apodemus flavicollis and A. uralensis Mice, Turkey
Figure 2
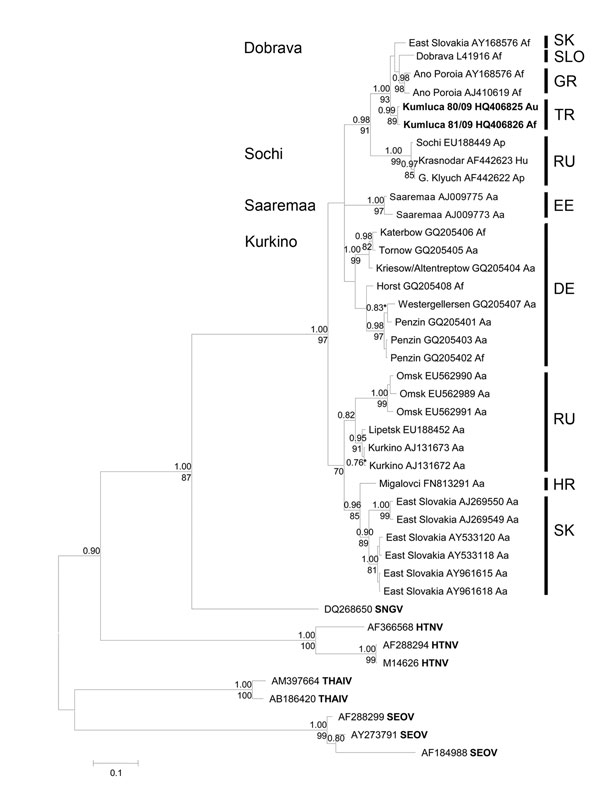
Figure 2. . Bayesian phylogenetic tree, based on an alignment of 450-nt long region of the small segment from various Dobrava-Belgrade virus lineages and other Murinae-associated hantaviruses. Posterior probabilities for Bayesian analysis are given under the branches and bootstrap values above the branches. Values lower than <0.7% and <70% are not shown. The sequences were aligned with ClustalW included in the BioEdit software package version 2.1 (http://www.mbio.ncsu.edu/bioedit/page2.html). The phylogenetic analyses were performed by using MrBayes 3.1.2 with Bayesian Metropolis-Hastings Markov Chain Monte Carlo (MCMC) tree-sampling methods based on 2 MCMC runs consisting of 4 chains of 2,000,000 with a burn-in of 25% and second by maximum-likelihood bootstrap analysis with 1,000 pseudoreplicates using MEGA5 (www.megasoftware.net). The Hasegawa-Kishino-Yano model with a discrete gamma distribution, to model evolutionary rate differences among sites (2 categories [+G, parameter = 0.8874]) according to jModeltest (http://code.google.com/p/jmodeltest2/) was used. Af, Apodemus flavicollis; Au, A. uralensis; Ap, A. ponticus; Hu, human; Aa, A. agrarius; SNGV, Sangassou virus; HTNV, Hantaan virus; THAIV, Thailand virus; SEOV, Seoul virus; SK, Slovakia; SLO, Slovenia; GR, Greece; TR, Turkey; RU, Russia; EE, Estonia (island Saaremaa); DE, Germany; HR, Croatia. Scale bar indicates number of nucleotide substitutions per site.