Volume 20, Number 10—October 2014
Letter
Usutu Virus in Bats, Germany, 2013
Figure
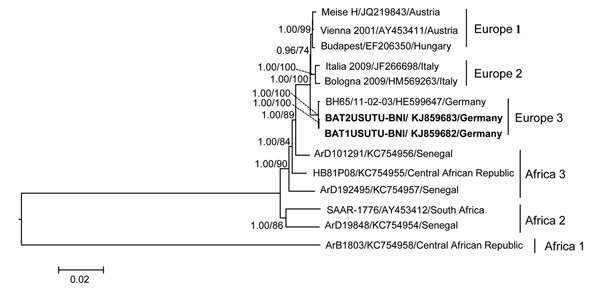
Figure. Maximum-likelihood phylogenetic tree of Pipistrellus bat Usutu viruses (USUV) based on full-length nucleotide sequences and showing the phylogenetic placement of the bat-derived USUV compared with human-, mosquito-, and bird-derived strains. The phylogenetic analyses were performed by using PhyML 3.0 (http://www.atgc-montpellier.fr/phyml/versions.php) with 1,000 pseudo-replicates and parallel Bayesian Markov chain Monte Carlo tree-sampling methods based on 2 runs consisting of 4 chains of 1,000,000 with a burn-in of 25% using MrBayes 3.1.2 (http://mrbayes.sourceforge.net/). The Akaike information criterion was chosen as the model selection framework and the general time-reversible model of sequence evolution with gamma-distributed rate variation among sites as the best model. Numbers at the nodes indicate maximum-likelihood bootstrap replicates (>70%) and parallel the posterior probability values (clade credibilities >90%). Boldface indicates USUV strains from Pipistrellus bats in Germany in 2013 (this study). Strain names, GenBank accession numbers, and countries of origin for sequences used to construct the tree are indicated on the branches. Scale bar indicates mean number of nucleotide substitutions per site.