Rickettsia spp. in Seabird Ticks from Western Indian Ocean Islands, 2011–2012
Muriel Dietrich

, Camille Lebarbenchon, Audrey Jaeger, Céline Le Rouzic, Matthieu Bastien, Erwan Lagadec, Karen D. McCoy, Hervé Pascalis, Matthieu Le Corre, Koussay Dellagi, and Pablo Tortosa
Author affiliations: Centre de Recherche et de Veille sur les Maladies Émergentes dans l’Océan Indien, Sainte Clotilde, Réunion Island, France (M. Dietrich, C. Lebarbenchon, C. Le Rouzic, M. Bastien, E. Lagadec, H. Pascalis, K. Dellagi, P. Tortosa); Université de La Réunion, Saint Denis, Réunion Island, France (M. Dietrich, C. Lebarbenchon, P. Tortosa, A. Jaeger, C. Le Rouzic, M. Bastien, M. Le Corre); Institut de Recherche pour le Développement, Sainte Clotilde (E. Lagadec, H. Pascalis, K. Dellagi); Maladies Infectieuses et Vecteurs: Ecologie, Génétique, Evolution et Contrôle, Montpellier, France (K.D. McCoy)
Main Article
Figure 3
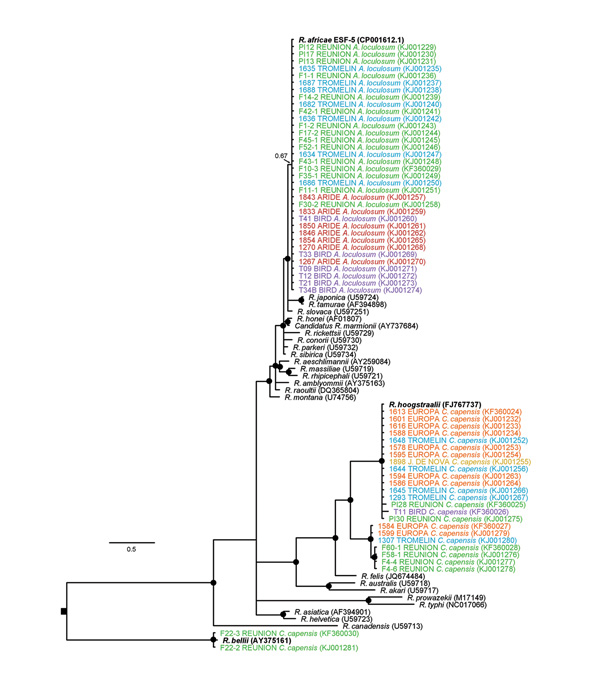
Figure 3. Maximum clade credibility tree for Rickettsia sppdetected in seabird ticks (Amblyomma loculosum and Carios capensis) of the western Indian Ocean as determined on the basis of a 913-bp fragment of the Rickettsia gltA geneThe nucleotide substitution model was selected by using the jModelTest 2.1.2 tool (https://code.google.com/p/jmodeltest2/), and Bayesian analyses were performed using MrBayes 3.1.2 (http://mrbayes.sourceforge.net/), with chain lengths of 2 million generations sampled every 1,000 generationsBlack dots indicate Bayesian posterior probabilities >0.7Taxa names are represented by the identification of the sample, the sampling site, and the tick speciesGenBank accession numbers are indicated in parenthesesThe sequences generated in this study are color-coded according to the geographic origin of samples (see Figure 1) and are accessible in GenBank (accession nosKF360024–KF360030 and KJ001229–KJ001281)Scale bars indicates nucleotide substitutions per site.
Main Article
Page created: April 18, 2014
Page updated: April 18, 2014
Page reviewed: April 18, 2014
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.