Volume 20, Number 6—June 2014
Letter
Novel Henipa-like Virus, Mojiang Paramyxovirus, in Rats, China, 2012
Cite This Article
Citation for Media
To the Editor: The genus Henipavirus (family Paramyxoviridae) contains 3 established species (Hendra virus, Nipah virus, and Cedar virus) and 19 newly identified species, including 1full-length sequenced virus, Bat Paramyxovirus Eidhel/GH-M74a/GHA/2009 (1,2). The zoonotic pathogens Hendra virus and Nipah virus have been associated with lethal neurologic and respiratory diseases in humans, horses, and pigs (3–5). The known natural reservoirs of henipaviruses are fruit bats (1,3); these viruses have not been reported in other wild animals. We report on a novel henipa-like virus, Mojiang paramyxovirus (MojV), in rats (Rattus flavipectus) in China.
In June 2012, in Mojiang Hani Autonomous County, Yunnan Province, China, severe pneumonia without a known cause was diagnosed in 3 persons who had been working in an abandoned mine; all 3 patients died. Half a year later, we investigated the presence of novel zoonotic pathogens in natural hosts in this cave. For the investigation, we collected anal swab samples from 20 bats (Rhinolophus ferrumequinum), 9 rats (R. flavipectus), and 5 musk shrews (Crocidura dracula) from the mine for virome analysis.
All samples were processed by using a virus particle–protected nucleic acid purification method, followed by sequence-independent PCR amplification of extracted RNA and DNA (6). The amplified viral nucleic acid libraries were then sequenced by using an Illumina Genome Analyzer II (Illumina Trading, Beijing, China) for a single read of 81 bp. All raw reads were then aligned to the nonredundant protein database of the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/RefSeq/) by using BLASTx (http://blast.ncbi.nlm.nih.gov/Blast.cgi) after filtering reads as described (6). The taxonomy of the aligned reads was parsed by using the MEGAN4 MetaGenome Analyzer (7).
On the basis of the nonredundant protein alignment results, we identified 38 sequence reads that were classified as Henipavirus spp. However, the sequences shared low nucleotide and amino acid identities with known henipaviruses. The reads were then used for reads-based PCR to identify the partial genome of this virus. The remaining genomic sequences were determined by using genome walking. The 5′ and 3′ untranslated regions were obtained by nested PCR with combined specific primers and henipavirus-specific degenerate primers as described (8), and the exact sequences of the 5′ and 3′ genome termini were determined by rapid amplification of cDNA ends.
MojV shares similar features with known henipaviruses. The virus has a genome length of 18,404 nt (submitted to GenBank under accession no. KF278639), and has the characteristic henipavirus gene order: 3′-nucleocapsid (N) protein (539 aa); P/V/W/C proteins (phosphoprotein; 694 aa, 464 aa, 434 aa, 177 aa); matrix protein (340 aa); fusion protein (545 aa); attachment glycoprotein (625 aa); and large (L) protein (2,277 aa)-5′ (Technical Appendix Figure). The predicted conserved sequences between genes showed features characteristic of henipaviruses (Technical Appendix Table). The central domain of the N protein contains 3 conserved motifs common in all paramyxoviruses: QXW [I/V] X3K [A/C] XT, FX2T[I/L][R/K]Φ[G/A][L/I/V]XT, and FX4YPX2ΦSΦAMG, where Φ is an aromatic amino acid (9). In addition, the RNA editing site (AAAAGG) for the processing of V and W proteins conserved in the phosphoprotein gene sequences of Hendra virus and Nipah virus was found, and 6 conserved domains within the L proteins of the order Mononegavirales (8) were found in the MojV L protein.
Figure
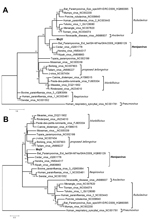
Figure. Phylogenetic trees based on the nucleocapsid proteins (A) and large proteins (B) of Mojiang paramyxovirus (MojV) and other previously reported paramyxovirusesBold font indicates MojV and Henipavirus sppScale bars indicate nucleotide substitutions...
The nucleotide identities of predicted MojV genes exhibited similarity with genes of known henipaviruses: N (53.0%–57.0% identity), phosphoprotein (37.8%–43.0% identity), matrix (59.5%–63.4% identity), fusion (47.5%–51.4% identity), attachment glycoprotein (36.6%–41.8% identity), and L (55.9%–58.6% identity) genes. Using MEGA5 (10), we used the phylogenetic trees based on N and L proteins to describe the evolutionary relationships between MojV and members of the family Paramyxoviridae (Figure). MojV clustered with the 4 members of the genus Henipavirus and was distant from other clusters. Thus, considering the similar genome features between MojV and other henipaviruses, we confirmed that MojV could be classified as a new species closely related to Henipavirus spp.
Specific nested primer sets targeting the L gene of MojV were designed to separately re-evaluate the 34 anal swab samples and some tissue samples. Of 9 anal swab samples from the R. flavipectus rats, 3 were positive for MojV, and a tissue sample from 1 of the 3 MojV-positive rats was also MojV positive (tissue was not collected from the other 2 rats). All 20 samples from R. ferrumequinum bats and all 5 samples from C. dracula musk shrews were MojV negative. The 3 MojV-positive anal swab samples were cultured in Vero E6, Hep2, and BHK21 cells for virus isolation; no cytopathic effects or viral replication was detected after 2 blind subculture passages.
Our study showed the presence of a rodent-origin, henipa-like virus, MojV, in China. R. flavipectus rats are the natural reservoir of MojV. This finding and its context indicate that Henipavirus spp. viruses might infect more mammalian hosts than previously thought and that bats may not be the only hosts of henipaviruses.
Acknowledgment
This work was supported by a National S&T Major Project (China Mega-Project for Infectious Disease; grant no. 2011ZX10004-001) from the People’s Republic of China, and by a Basic Research and Operating Expenses grant (no. 2013IPB301) from the Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College.
References
- Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Bats host major mammalian paramyxoviruses. Nat Commun. 2012;3:796.PubMedGoogle Scholar
- Marsh GA, de Jong C, Barr JA, Tachedjian M, Smith C, Middleton D, Cedar virus: a novel henipavirus isolated from Australian bats. PLoS Pathog. 2012;8:e1002836. DOIPubMedGoogle Scholar
- Smith I, Wang LF. Bats and their virome: an important source of emerging viruses capable of infecting humans. Curr Opin Virol. 2013;3:84–91.
- Mendez DH, Judd J, Speare R. Unexpected result of Hendra virus outbreaks for veterinarians, Queensland, Australia. Emerg Infect Dis. 2012;18:83–5. DOIPubMedGoogle Scholar
- Sazzad HM, Hossain MJ, Gurley ES, Ameen KM, Parveen S, Islam MS, Nipah virus infection outbreak with nosocomial and corpse-to-human transmission, Bangladesh. Emerg Infect Dis. 2013;19:210–7. DOIPubMedGoogle Scholar
- Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol. 2012;86:10999–1012. DOIPubMedGoogle Scholar
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–60. DOIPubMedGoogle Scholar
- Miller PJ, Boyle DB, Eaton BT, Wang LF. Full-length genome sequence of Mossman virus, a novel paramyxovirus isolated from rodents in Australia. Virology. 2003;317:330–44. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Wong BH, Wong AY, Tsoi HW, Wang M, Identification and complete genome analysis of three novel paramyxoviruses, Tuhoko virus 1, 2 and 3, in fruit bats from China. Virology. 2010;404:106–16. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
Figure
Cite This Article1These authors contributed equally to this article.
Related Links
Table of Contents – Volume 20, Number 6—June 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Qi Jin, No. 6 Rongjing East St, Yizhuang, Beijing, 100176, China
Top