Dengue Virus Type 3, South Pacific Islands, 2013
Van-Mai Cao-Lormeau

, Claudine Roche, Didier Musso, Henri-Pierre Mallet, Tenneth Dalipanda, Alfred Dofai, Francisco Nogareda, Eric J. Nilles, and John Aaskov
Author affiliations: Institut Louis Malardé, Papeete, Tahiti, French Polynesia (V.-M. Cao-Lormeau, C. Roche, D. Musso); Direction de la Santé, Papeete (H.-P. Mallet); Ministry of Health and Medical Services, Honiara, Solomon Islands (T. Dalipanda); National Referral Hospital, Honiara (A. Dofai); World Health Organization, Suva, Fiji (F. Nogareda, E.J. Nilles); Queensland University of Technology, Brisbane, Queensland, Australia (J. Aaskov)
Main Article
Figure 2
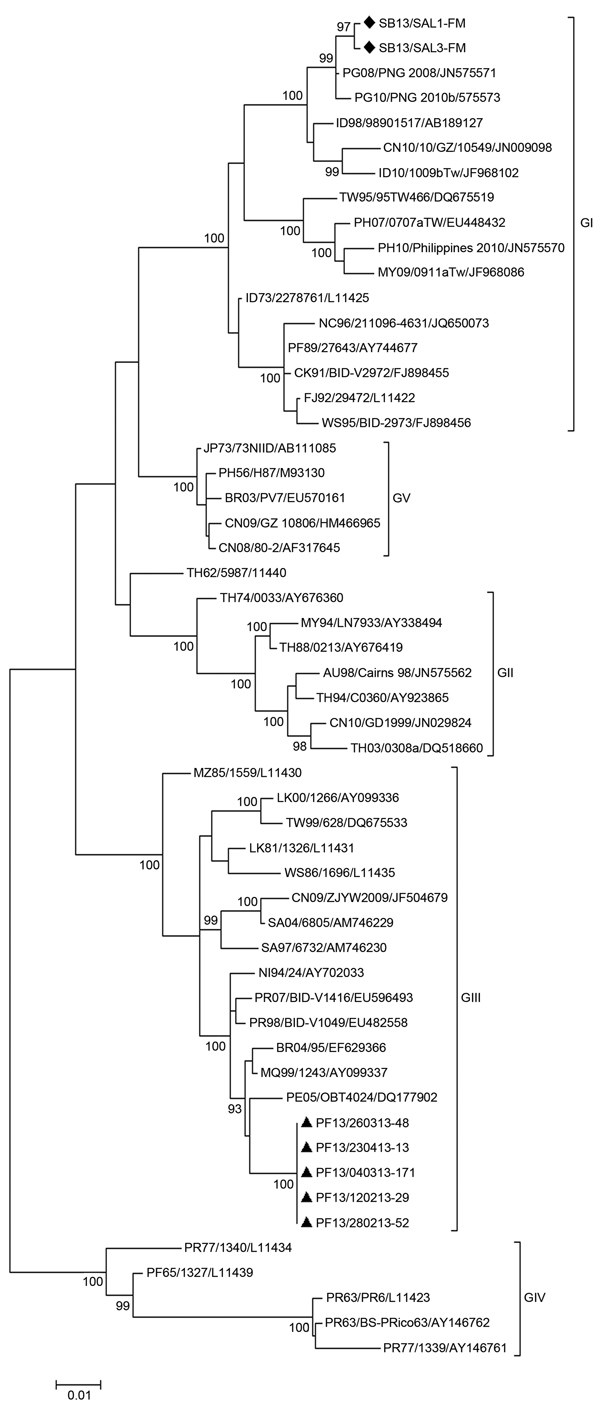
Figure 2. Evolutionary relationships of 54 dengue virus type 3 (DENV-3) gene sequencesThe phylogenetic tree was obtained by using the maximum-likelihood method based on the Kimura 2-parameter model and MEGA 5 software (http://www.megasoftware.net/)The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown for values >90Each strain is labeled by country of origin/strain name/GenBank accession number (if available)Black diamonds indicate DENV-3 strains sequenced in this study that were isolated during the DENV-3 epidemic in the Solomon Islands in 2013Black triangles indicate DENV-3 strains sequenced in this study that were isolated during the DENV-3 epidemic in French Polynesia in 2013Genotypes are indicated on the right side of the treeScale bar indicates nucleotide substitutions per site.
Main Article
Page created: May 16, 2014
Page updated: May 16, 2014
Page reviewed: May 16, 2014
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.