Volume 22, Number 11—November 2016
Dispatch
Mayaro Virus in Child with Acute Febrile Illness, Haiti, 2015
John Lednicky, Valery Madsen Beau De Rochars, Maha Elbadry, Julia Loeb, Taina Telisma, Sonese Chavannes, Gina Anilis, Eleonora Cella, Massimo Ciccozzi, Bernard Okech, Marco Salemi, and J. Glenn Morris
Figure
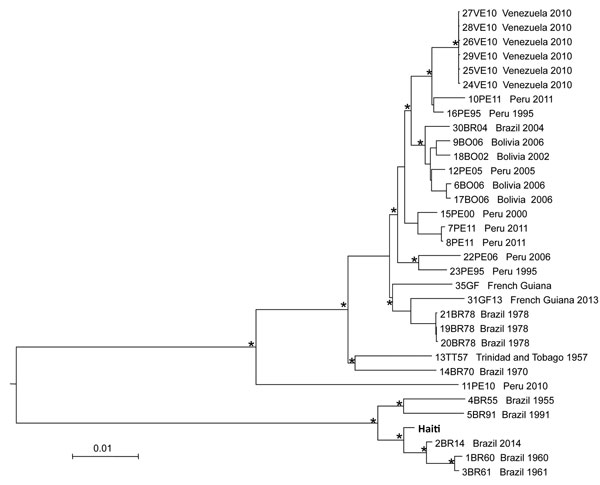
Figure. Neighbor-joining tree of full-genome Mayaro virus sequences. The tree was inferred from pairwise distances estimated with the best fitting nucleotide substitution model (general time reversible plus gamma). The tree includes the isolate from Haiti identified in this study (in boldface) and all full-genome sequences with known country of origin and sampling date downloaded from GenBank. Branches are drawn according to the scale bar at the bottom, which indicates nucleotide substitutions per site. An asterisk along a branch indicates bootstrap support >90% for the subtending clade.
Page created: March 10, 2017
Page updated: March 10, 2017
Page reviewed: March 10, 2017
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.