Volume 22, Number 3—March 2016
Dispatch
Far East Scarlet-Like Fever Caused by a Few Related Genotypes of Yersinia pseudotuberculosis, Russia
Cite This Article
Citation for Media
Abstract
We used multivirulence locus sequence typing to analyze 68 Yersinia pseudotuberculosis isolated in Russia during 1973–2014, including 41 isolates from patients with Far East scarlet-like fever. Four genotypes were found responsible, with 1 being especially prevalent. Evolutionary analysis suggests that epidemiologic advantages could cause this genotype’s dominance.
Far East scarlet-like fever (FESLF), a rare and poorly studied disease caused by Yersinia pseudotuberculosis, was first described in 1959, when an outbreak involving >300 hospitalized patients occurred in the city of Vladivostok, Russia, on the coast of the Pacific Ocean (1). Since the 1960s, multiple outbreaks and sporadic cases of FESLF, mainly associated with consumption of contaminated vegetables, have been reported from far eastern and northern parts of Russia and other countries in Eurasia (2–4).
Comparing clinical patterns of FESLF and pseudotuberculosis showed that FESLF is not just a form of pseudotuberculosis but is an independent infectious disease that was unknown until the 1960s (4). FESLF is an acute disease with a cyclic course that includes severe fever and early signs such as rash that covers the body, particularly the face, neck, toes, and hands; these signs have become known as “hood,” “gloves,” and “socks” (Technical Appendix Figure 1). Typical features of FESLF include a “raspberry tongue” and well-defined nipples. Erythema nodosum can occur with relapse; lamellar or defurfuration on earlobes, hands, palms, feet, and trunk appears during the recovery period. We sought to determine clonal relationships of Y. pseudotuberculosis strains responsible for cases of FESLF reported in Russia during 1973–2014 and environmental strains found in vegetables and small rodents.
Figure 2
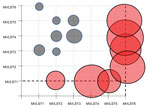
Figure 2. Graphic representation of the evolutionary analysis that tested the hypothesis of equality of evolutionary rates between multivirulence locus sequence type (MVLST) genotypes for study of Far East scarlet-like fever caused by...
Our study examined 68 isolates collected in Russia during 1973–2014, including 17 outbreak and 24 sporadic isolates from humans and 15 rodent and 11 vegetable isolates (Technical Appendix Table 1, Figure 2). All but 3 isolates belonged to the O1b serotype; these 3 isolates belonged to the O3 serotype. The most recent FESLF isolates (from 2014) came from a patient who showed typical signs of FESLF, including a cyclic course, fever, and “raspberry tongue.” A comparison of clinical signs and symptoms in historical versus recent patients suggested that the disease had not evolved since its first description.
The isolates were kept frozen until the experiment started. To characterize clonal relationships of the strains, we applied the multilocus sequence typing (MLST) scheme developed by Laukkanen-Ninios et al. (5). PCR products were obtained with primers and conditions listed at the Yersinia pseudotuberculosis MLST database (University of Warwick, Coventry, UK; http://mlst.ucc.ie/mlst/dbs/Ypseudotuberculosis).
We found 3 MLSTs among FESLF isolates: MLST2 (n = 33), MLST26 (n = 5), and MLST32 (n = 3); this MLST was specific for serotype O3 (Table 1). All but 1 vegetable isolate belonged to MLST2, which was also found in 9 (60%) of 15 rodent isolates. MLST2 prevailed among isolates from all sources.
MLST analysis was complemented with sequencing of 4 virulence genes involved in critical steps of generalized infection: intestine barrier crossing (inv and yadA) (6,7) and macrophage activity regulation (yopE and cnf) (8,9) (Tables 1, 2). The genes inv and cnf are chromosomal, whereas yopE and yadA are encoded on the virulence plasmid of Yersinia (pYV). Sequences from this study have been deposited into GenBank (accession nos. KR028003–KR028011). A total of 4 distinct virulence sequence types (VSTs) were found (Table 1).
Figure 1
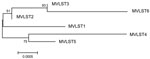
Figure 1. Maximum-likelihood tree generated with concatenated multivirulence locus sequence type (MVLST) sequences for study of Far East scarlet-like fever caused by a clonal group of Yersinia pseudotuberculosis, Russia. Reliability values for the...
Combining MLST and VST gave rise to 6 multivirulence locus sequence types (MVLSTs) (Table 1). The sequences of 10 MVLST genes (excluding cnf) were used to build a maximum-likelihood tree with MEGA6 (10). We excluded the cnf gene from the analysis because the dominant allele carries a nonsense mutation that interrupts the polypeptide after Asn181. The maximum-likelihood tree divided into 2 subclades (Figure 1). One subclade united MVLSTs found in FESLF isolates and MVLST6, which was found only in rodent isolates. The second subclade united MVLSTs found in rodent and vegetable isolates.
The diversity of virulence genes was analyzed with DnaSP software version 5.10 (11; Table 2). A noticeable feature of virulence genes was the predominance of nonsynonymous substitutions, whereas basic parameters of nucleotide diversity were similar in virulence and housekeeping genes (Table 2). Positive selection was confirmed for yopE by the Tajima neutrality test implemented in MEGA6. The diversity was especially low among strains from the FESLF subcluster. MVLST1 and MVLST2 shared the VST1 type (Table 1). MVLST3 shared VST2 with MVLST6 found in rodent strains.
Plasmids, particularly the pYV plasmid, are central to the virulence of Yersiniae (12). The pYV-specific markers yopE and yadA were found in all strains. The presence of the additional plasmid pVM82/pYpsIP31758.1 was screened with PCR specific to the dotA gene (3), which was found in all but 4 MVLST1 strains but not in other genotypes. Plasmid purification (13) confirmed results obtained from PCR-based screening (data not shown). An additional small plasmid was found in MVLST1 strains that lacked pVM82/pYpsIP31758.1. Consequently, plasmid profiling divided MVLST1 into 2 subtypes, MVLST1a and MVLST1b, without changing other MVLSTs (Table 1).
Our findings show that FESLF clinical manifestations are caused by strains belonging to at least 4 distinct genotypes, with predominance of MVLST1a (MLST2/VST1/pVM82). We consider the MVLST1a genotype to be generally dominant among strains responsible for FESLF in Russia, a suggestion supported by the finding that MVLST1a appears to be the only genotype that carries the pVM82/pYpsIP31758.1 plasmid. A body of epidemiologic data has shown that most epidemic and many sporadic FESLF strains carry this plasmid (3,13).
The fact that full FESLF symptomatology is caused by several distinct genotypes supports the view that specific virulence traits are characteristic of FESLF-associated strains (2,3) and suggests that the dominance of the MVLST1a genotype could be caused by its epidemiologic advantages rather than its pathogenic traits. The prevalence of MVLST1a among all isolate sources suggests the genotype’s wider dissemination in the region we studied, which supports the possibility that this clone has epidemiologic advantages.
To further address this question, we used an evolutionary analysis implemented in MEGA6 (10) to test the hypothesis of equality of evolutionary rates by using the χ2 test for pairwise comparison of concatenated sequences of MVLST markers, with the Y. pestis sequence used as an outgroup. The hypothesis of equal rates between MVLST1 and other genotypes was rejected (p<0.05; Figure 2). The molecular clock test performed with MEGA6 by comparing the maximum-likelihood values with and without molecular clock constraints under the Tamura-Nei model supported this conclusion. The inequality of evolutionary rates favors the idea of more effective reproduction and growth of MVLST1 strains in the environment, possibly because of better adaptation to environmental niches. Another clone with divergent evolutionary rates was the rare MVLST6 (MLST64/VST2) genotype, which has been isolated from small rodents in the Far East of Russia (i.e., in this study and according to data on the isolation of MlST64, listed in the Y. pseudotuberculosis MLST database).
FESLF, a relatively new disease, is caused by the bacterium that evolved into the causative agent of plague (14). The evolution of Y. pestis is linked to loss of functionality of some factors that are active in Y. pseudotuberculosis and to the acquisition of additional factors of both plasmid and chromosomal origin; these alterations enable the organism to adapt and occupy new environmental niches (14). The FESLF causative agent lost at least 2 chromosomally encoded virulence loci, cnf and HPI; its most successful clone, MVLST1a, acquired an additional plasmid. The geographic region where the first outbreaks of FESLF were registered seems close, if not identical, to the region where Y. pestis emerged. Overall, our data support the view of Y. pseudotuberculosis as a rapidly developing pathogenic species, whereas its wide dissemination in the environment promotes selection of clones that are potentially hazardous for humans (2–4,15).
Dr. Timchenko is a leading scientist at the Laboratory of Molecular Epidemiology and Microbiology at the Institute of Epidemiology and Microbiology named after G.P. Somov, Vladivostok, Russia. Her research interests include microbiology and pathogenic traits of Yersinia pseudotuberculosis.
Dr. Ermolaeva is a head of the Laboratory of Ecology of Pathogenic Bacteria at Gamaleya Institute of Epidemiology and Microbiology, Moscow, Russia. Her main research interests are in the field of molecular ecology of pathogenic bacteria.
Acknowledgment
We thank Margarita Kurnaeva for help with multivirulence locus sequence typing experiments. We also thank Biomedes Ltd. for assistance with English grammar improvements in the manuscript.
References
- Grunin II, Somov GP, Zalmover IIu. Far Eastern scarlatinoid fever [in Russian]. Voen Med Zh. 1960;
8 :62–6.PubMedGoogle Scholar - Fukushima H, Matsuda Y, Seki R, Tsubokura M, Takeda N, Shubin FN, Geographical heterogeneity between far eastern and western countries in prevalence of the virulence plasmid, the superantigen Yersinia pseudotuberculosis-derived mitogen, and the high-pathogenicity island among Yersinia pseudotuberculosis strains. J Clin Microbiol. 2001;39:3541–7. DOIPubMedGoogle Scholar
- Eppinger M, Rosovitz MJ, Fricke WF, Rasko DA, Kokorina G, Fayolle C, The complete genome sequence of Yersinia pseudotuberculosis IP31758, the causative agent of Far East scarlet-like fever. PLoS Genet. 2007;3:e142. DOIPubMedGoogle Scholar
- Somov G. Far-East scarlet-like fever. Moscow: Medicine; 1979.
- Laukkanen-Ninios R, Didelot X, Jolley KA, Morelli G, Sangal V, Kristo P, Population structure of the Yersinia pseudotuberculosis complex according to multilocus sequence typing. Environ Microbiol. 2011;13:3114–27. DOIPubMedGoogle Scholar
- Eitel J, Dersch P. The YadA protein of Yersinia pseudotuberculosis mediates high-efficiency uptake into human cells under environmental conditions in which invasin is repressed. Infect Immun. 2002;70:4880–91. DOIPubMedGoogle Scholar
- Isberg RR, Voorhis DL, Falkow S. Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell. 1987;50:769–78. DOIPubMedGoogle Scholar
- Hoffmann C, Pop M, Leemhuis J, Schirmer J, Aktories K, Schmidt G. The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J Biol Chem. 2004;279:16026–32. DOIPubMedGoogle Scholar
- Wang X, Parashar K, Sitaram A, Bliska JB. The GAP activity of type III effector YopE triggers killing of Yersinia in macrophages. PLoS Pathog. 2014;10:e1004346. DOIPubMedGoogle Scholar
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. DOIPubMedGoogle Scholar
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. DOIPubMedGoogle Scholar
- Portnoy DA, Martinez RJ. Role of a plasmid in the pathogenicity of Yersinia species. Curr Top Microbiol Immunol. 1985;118:29–51. DOIPubMedGoogle Scholar
- Shubin FN, Gintsburg AL, Kitaev VM, Ianishevskiĭ NV, Zenkova ZG. Analysis of the plasmid composition of Yersinia pseudotuberculosis strains and its use for typing pseudotuberculosis pathogens. Mol Gen Mikrobiol Virusol. 1989;6:20–5.PubMedGoogle Scholar
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A. 1999;96:14043–8. DOIPubMedGoogle Scholar
- Wren BW. The Yersiniae—a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol. 2003;1:55–64. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors were co–principal investigators.
Table of Contents – Volume 22, Number 3—March 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Svetlana A. Ermolaeva, Gamaleya Institute of Epidemiology and Microbiology, Gamaleya St 18, Moscow 123098, Russia
Top