Volume 22, Number 4—April 2016
Dispatch
Deletion Variants of Middle East Respiratory Syndrome Coronavirus from Humans, Jordan, 2015
Cite This Article
Citation for Media
Abstract
We characterized Middle East respiratory syndrome coronaviruses from a hospital outbreak in Jordan in 2015. The viruses from Jordan were highly similar to isolates from Riyadh, Saudi Arabia, except for deletions in open reading frames 4a and 3. Transmissibility and pathogenicity of this strain remains to be determined.
Middle East respiratory syndrome coronavirus (MERS-CoV) was first recognized in 2012 as the cause of severe lower respiratory tract infection in humans (1). As of November 13, 2015, a total of 1,618 cases of laboratory-confirmed MERS-CoV infection and 579 associated deaths had been reported to the World Health Organization (2). Human-to-human transmission of MERS-CoV can occur in households and hospitals, but thus far, major genomic changes associated with host switching, as have occurred with severe acute respiratory syndrome (SARS) CoV, have not been noted for MERS-CoV (3–9). We characterized MERS-CoVs from a recent hospital outbreak in Jordan.
During August and September 2015, an outbreak of MERS-CoV infection occurred in hospitals in Jordan (10). MERS-CoV–positive throat swab or bronchiolar lavage samples were obtained from each patient; of these, 13 samples were labeled Jordan-1-2015 through Jordan-13-2015 and shipped on dry ice to Erasmus University Medical Center (Rotterdam, the Netherlands) for testing. Total RNA was isolated from 140 μL of each sample by use of a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) and quantified by TaqMan assay targeting the envelope gene, as described previously (11). Full MERS-CoV genome sequencing was performed on the sample with the highest viral load, obtained on August 24, 2015, from a 60-year-old man who died of the disease 3 days later. At the end of July 2015, this man had traveled from Jeddah, Saudi Arabia, to Jordan for his annual vacation. From the throat swab sample from this man, 76,082 sequence reads—of which 851 were specific for MERS-CoV—were obtained by using 454 deep-sequencing (12), revealing ≈85% of the MERS-CoV genome with coverage of 1–178 reads at single-nucleotide positions. Missing sequences and low-coverage regions were obtained by conventional Sanger sequencing, except for 6 nt at the 5′ end.
Figure 1
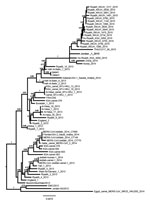
Figure 1. Phylogenetic analysis of Middle East respiratory syndrome coronavirus (MERS-CoV) isolated from Jordan (Jordan-1-2015; boldface) compared with reference strains. Genome sequences of representative isolates were aligned by using ClustalW, and a phylogenetic...
Figure 2
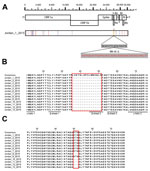
Figure 2. Genomic characterization of Middle East respiratory syndrome coronaviruses (MERS-CoVs) from Jordan. A) Nucleotide differences between MERS-CoV strain Jordan-1-2015 and the consensus sequence of the Riyadh 2015 cluster. Nucleotide changes are indicated...
Phylogenetic analysis of the complete viral genome of this sample revealed that this virus, tentatively called Jordan-1-2015, clusters with viruses detected in humans in Riyadh in 2015 (Figure 1). Alignment of the full genomic sequences of this virus with the Riyadh 2015 isolates showed that they were >99.7% similar. Amino acids in the spike protein were the same in Jordan-1-2015 and the consensus sequence of the Riyadh 2015 viruses. Of note, Jordan-1-2015 contained a 48-nt (16 aa) in-frame deletion in open reading frame (ORF) 4a that had not been detected in any other MERS-CoV (Figure 2, panel A). This ORF is present only in lineage 2c betacoronaviruses and encodes protein 4a, which has recently been shown to inhibit type I interferon production, presumably by binding and masking double-stranded RNA from detection by RNA sensors such as RIG-1 (retinoic acid–inducible gene 1)–like helicases and the double-stranded RNA-binding protein PACT (protein activator of the interferon-induced protein kinase) (13,14). Subsequent sequence analysis showed that all 13 viruses from patients sampled during this outbreak contained the deletion and that the flanking regions were identical, indicating that this deletion mutant was transmitted (Figure 2, panel B).
To further characterize the viruses in the clinical samples, we subjected all samples to titration on Vero cells (American Type Culture Collection no. CCL-81). We observed cytopathic changes in cells inoculated with samples Jordan-1-2015 and Jordan-10-2015 (obtained on September 17 from a 29-year-old man, who survived the illness); however, no cytopathic changes were observed in the cells inoculated with the other samples. Subsequently, we obtained complete genomes by deep sequencing of the second-passage viruses, with high coverage of reads at single-nucleotide positions. No cell culture adaptive mutations in the genome sequence were observed for the virus isolated from clinical sample Jordan-1-2015. Pairwise comparison of Jordan-1-2015 with Jordan-10-2015 revealed that both viruses were highly similar (99.99% nt identity) and confirmed that both carried the 48-nt deletion in ORF4a. However, in addition to this deletion, a 9-nt (3-aa) in-frame deletion was observed in the genome of Jordan-10-2015. This deletion was located in ORF3, which encodes a protein with an unknown function. The presence of this deletion in the virus from clinical sample Jordan-10-2015 was confirmed by Sanger sequencing, excluding the possibility that it was an adaptation to cell culture. Next, sequencing of ORF3 from all other clinical samples revealed that the ORF3 deletion was present in a subgroup of clinical samples (Figure 2, panel C). These samples had been collected toward the end of the outbreak, suggesting that the ORF3 deletion may be an adaptation resulting from sustained transmission between humans. All sequences obtained in this study were deposited in GenBank (accession nos. KU233362–KU233377). Identical deletions in ORF3 and ORF4a in the same sample set from Jordan were independently confirmed by Erasmus University Medical Center and the Centers for Disease Control and Prevention (Atlanta, GA, USA).
Despite the human-to-human transmission capacity of MERS-CoV and substantial sequencing efforts, major genomic changes associated with human adaptation have not been described (3–8). We observed a large-scale deletion in ORF4a and a small deletion in ORF3 in MERS-CoVs that caused the outbreak in Jordan.
One of the most striking genomic changes observed in SARS-CoV isolated from humans soon after its transmission from animals was the acquisition of a 29-nt deletion in ORF8, an accessory protein with an unresolved function (9). The SARS-CoV ORF8 deletion disrupted the reading frame of the early-stage human isolates and created 2 new ORFs, designated ORF8a and ORF8b. Although no clear roles have been designated for both ORFs, viruses containing this deletion predominated later during the epidemic, showing that these viruses were able to spread efficiently from human to human.
The deletion we describe in ORF4a of MERS-CoV does not result in the removal of an entire gene, because the reading frame is not disrupted, suggesting that this mutation is not a loss-of-function mutation. However, whether this ORF4a deletion variant can still bind double-stranded RNA needs to be assessed because the deleted region contains a predicted β-sheet belonging to the classical double-stranded RNA binding αβββα fold of this protein (13; Figure 2, panel B). The data from this study indicate that adaptive pressures possibly exerted by the human host may operate on the ORF4a and ORF3 regions. Alternatively, proteins encoded by these ORFs could have functions specific for the camel host, causing them to be redundant in humans and enabling accumulation of mutations that do not affect viral fitness in humans. Future studies of the roles of these proteins are needed.
The deletion located in the type I interferon antagonist protein 4a was detected in all samples collected during the outbreak. Another small-scale deletion was detected in ORF3 in a subset of samples collected later in the outbreak. Overall, the data suggest that MERS-CoV ORF4a and ORF3 can acquire deletions by selection or by chance. Whether these deletions affect the transmissibility or pathogenicity of this particular MERS-CoV strain needs to be addressed. The finding that all viruses analyzed contained the ORF4a deletion suggests that all patients were infected with the same virus. Although unlikely, some MERS patients may have indirectly become infected through independent transmission of viruses in dromedaries carrying highly related viruses, some of which acquired deletions in their genome. Therefore, a more detailed epidemiologic investigation of this outbreak is needed. These data underscore the need for close monitoring of the molecular evolution of MERS-CoV.
Mr. Lamers is a PhD student at the Department of Viroscience, Erasmus University Medical Center, Rotterdam, the Netherlands. His research interests include the molecular and functional characterization of emerging viruses.
Acknowledgment
This study was supported by ZonMW, TOP Project (grant no. 91213066), EU H2020 COMPARE (grant agreement no. 643476), and the Erasmus Graduate Program Infection and Immunity (NWO grant no.022.005.032).
References
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–20. DOIPubMedGoogle Scholar
- World Health Organization. Emergencies preparedness, response: Middle East respiratory syndrome coronavirus (MERS-CoV) maps and epicurves [cited 2015 Nov 18]. http://www.who.int/csr/disease/coronavirus_infections/maps-epicurves/
- Drosten C, Meyer B, Müller MA, Corman VM, Al-Masri M, Hossain R, Transmission of MERS-coronavirus in household contacts. N Engl J Med. 2014;371:828–35. DOIPubMedGoogle Scholar
- Assiri A, McGeer A, Perl TM, Price CS, Al Rabeeah AA, Cummings DA, Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med. 2013;369:407–16. DOIPubMedGoogle Scholar
- Cotten M, Watson SJ, Kellam P, Al-Rabeeah AA, Makhdoom HQ, Assiri A, Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. 2013;382:1993–2002. DOIPubMedGoogle Scholar
- Drosten C, Muth D, Corman VM, Hussain R, Al Masri M. HajOmar W, et al. An observational, laboratory-based study of outbreaks of Middle East respiratory syndrome coronavirus in Jeddah and Riyadh, Kingdom of Saudi Arabia, 2014. Clin Infect Dis. 2015;60:369–77.
- Fagbo SF, Skakni L, Chu DKW, Garbati MA, Joseph M, Peiris M, Molecular epidemiology of hospital outbreak of Middle East respiratory syndrome, Riyadh, Saudi Arabia, 2014. Emerg Infect Dis [cited 2015 Oct 27]. DOIGoogle Scholar
- Seong MW, Kim SY, Corman VM, Kim TS, Cho SI, Kim MJ, Microevolution of outbreak-associated Middle East respiratory syndrome coronavirus, South Korea, 2015. Emerg Infect Dis [cited 2015 Nov 29]. DOIGoogle Scholar
- Chinese SARS Molecular Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–9. DOIPubMedGoogle Scholar
- World Health Organization. Global alert and response (GAR). Middle East respiratory syndrome coronavirus (MERS-CoV)—Jordan [cited 2015 Nov 18]. http://www.who.int/csr/don/01-september-2015-mers-jordan/en/
- Haagmans BL, Al Dhahiry SH, Reusken CB, Raj VS, Galiano M, Myers R, Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis. 2014;14:140–5 and. DOIPubMedGoogle Scholar
- van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio. 2012;3:e00473–12. DOIPubMedGoogle Scholar
- Siu KL, Yeung ML, Kok KH, Yuen KS, Kew C, Lui PY, Middle East respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J Virol. 2014;88:4866–76. DOIPubMedGoogle Scholar
- Niemeyer D, Zillinger T, Muth D, Zielecki F, Horvath G, Suliman T, Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J Virol. 2013;87:12489–95. DOIPubMedGoogle Scholar
Figures
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 22, Number 4—April 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Bart L. Haagmans, Department of Viroscience, Erasmus MC, Wytemaweg 80, 3015 CN, Rotterdam, the Netherlands; ; Aktham Haddadin, National Influenza Centre, CPHL, Laboratory Directorate, Jordan Ministry of Health, Amman, Jordan
Top