Volume 22, Number 6—June 2016
Dispatch
Whole-Genome Analysis of Cryptococcus gattii, Southeastern United States
Cite This Article
Citation for Media
Abstract
Cryptococcus gattii is a recognized pathogenic fungus along the Pacific coast of the United States from California to Washington. Here we report that C. gattii may also be endemic to the southeastern United States and has probably been present there longer than in the Pacific Northwest.
The pathogenic fungus Cryptococcus gattii, recently designated as a separate species from Cryptococcus neoformans, is now recognized as a separate species complex (1–3). In the United States, C. gattii was first identified in culture collections in the late 1960s, when a substantial proportion of C. neoformans isolates from California were identified as serotypes B or C (now C. gattii serotypes A and B) (4,5). Since then, C. gattii has been identified not only in California but also in the Pacific Northwest (PNW) states of Washington and Oregon, where it is now considered endemic (6). Although non–travel-associated cases of infection, typically manifesting as pneumonia or meningitis, are occasionally reported in other areas of the United States (7,8), C. gattii has not been considered to be endemic to any other areas of the United States.
Figure 1

Figure 1. Maximum-parsimony tree of multilocus sequence typing analysis of VGI isolates of Cryptococcus gattii from the southeastern United States. In the predominant clade, 1 isolate was from Michigan; all remaining isolates were...
The Centers for Disease Control and Prevention (CDC; Atlanta, GA, USA) has been performing passive surveillance for C. gattii in the United States since 2009. Isolates were collected as part of routine surveillance for C. gattii or submitted to the Fungal Reference Laboratory at the CDC for identification. We identified all isolates to the species level as described (8) and used the consensus protocol for multilocus sequence typing (MLST) (9). In the course of this surveillance, several isolates were received from states in the southeast; genotypic analysis revealed that these isolates consisted of 2 molecular types, VGI and VGIII (8). Of >400 isolates received from throughout the United States, 42 were molecular type VGI. MLST analysis of VGI isolates showed a cluster of isolates with a single MLST sequence type originating in the southeastern states (Figure 1). Five isolates from Georgia and 1 each from Florida, Tennessee, and Michigan were indistinguishable by MLST (sequence type 162). A second isolate from Georgia differed from this group by 1 single-nucleotide polymorphism (SNP) in the intergenic spacer gene locus.
Figure 2
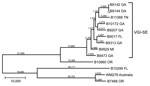
Figure 2. Maximum-parsimony tree of whole-genome sequence data of isolates of Cryptococcus gattii from the southeastern United States. All bootstrap values were 100%. Numbers on branches are SNPs. Nearest neighbor isolates were included...
Because the population of VGI isolates from the southeastern states (VGI-SE) was only recently identified, it might be a newly emerging population similar to the independent emergences of VGIIa and VGIIc in the PNW (6–8). To further characterize this cluster, we used whole-genome sequence typing (WGST) of isolates from this cluster, other closely related isolates, and unrelated control isolates (Table 1; Figure 2). We sequenced isolates using Illumina HiSeq or MiSeq technology (Illumina Inc., San Diego, CA, USA), used GATK Unified Genotyper version 2.4 (Broad Institute, Cambridge, MA, USA) for SNP detection, and constructed maximum-parsimony SNP trees using PAUP* (Sinauer Associates, Inc., Sunderland, MA, USA), as previously described (6).
Although the overall topology of the tree we generated remained almost identical to that seen by MLST, we were able to separate each of the isolates within the cluster using WGST (Figure 2). A total of 41,024 SNPs were within the VGI-SE cluster, which had an average branch length of 4,558 SNP differences between any 2 isolates. The isolate from Georgia that differed in MLST by 1 SNP was placed well within the cluster by WGST. Outside the cluster, the nearest neighbor, an isolate collected from a resident of Oregon with a history of living near the southeastern United States in Texas (Table 1), differed by >29,000 SNPs. The other cluster of VGI isolates was separated from VGI-SE by 49,992 SNPs.
Previously, whole-genome sequencing was performed on isolates of C. gattii molecular type VGII exclusively from the PNW. Results showed that the VGII isolates consisted of 3 different highly clonal and recently emerged populations with only 107, 132, and 137 SNPs identified within the VGIIa, VGIIb, and VGIIc populations, respectively (10) (Table 2). This finding indicated very recent divergence within each of these 3 subtypes (8). The average branch length between any 2 isolates was <18 SNPs for the VGIIa and VGIIc populations and only slightly higher for the VGIIb population. On the basis of SNP diversity, the VGI-SE subtype is substantially older than the clonal VGII populations in the PNW and has likely been in the United States for a much longer time.
Two of the isolates from Georgia that we analyzed differed by only 14 SNPs. These isolates, accessed from the same national reference laboratory, were submitted without clinical history; therefore, we cannot discern whether they represent the diversity of 2 isolates from 1 patient over time or whether they are part of a local clonal population.
All of the patients for whom disease manifestation was known had meningitis (Table 1). This finding differs notably from C. gattii VGII patients in the PNW, among whom a substantial proportion (59%) had a primary pulmonary infection (8). The average age of the patients with isolates in the VGI-SE cluster was 46 (range 18–70) years. For most patients, the source of infection was unknown, although 2 of the patients had been exposed to rotting wooden structures (1 demolished a wooden structure, 1 power-washed old houses).
Our data indicate that the clonal diversity of the C. gattii VGI-SE clade in the southeastern United States is an order of magnitude greater than that seen in isolates from the PNW. Gillece et al. hypothesized that the emergence in the PNW began in the mid-1990s in British Columbia, Canada, and spread to the United States in the early 2000s (12). Comparison of the average number of SNPs detected among the 3 VGII clonal groups in the PNW with the average number of SNPs detected between the VGI-SE isolates indicates that the VGI-SE isolates have been in place substantially longer. The limitation to this conclusion is that we do not know the rate of mutation for the VGI or VGII molecular types; however, there is no reason to believe that they would be substantially different. There may also be higher rate of recombination within the VGI-SE clade related to opposite-sex or same-gender sexual recombination. The isolates from this clade for which the mating type is known are all α, but that does not preclude the possibility that some mating type a isolates exist in the environment in the southeastern United States.
The first identified isolate of C. gattii in the United States was reported from West Virginia in 1924 (11), although the isolate was not recognized as C. gattii VGI until decades later (12). Two other historical reports of C. gattii isolates from the southeastern United States exist: the first, from 1968, describes clinical and environmental isolates from Savannah, Georgia; the other, from 1982, describes isolates from Alabama, Tennessee, and Louisiana from patients with no travel history to a C. gattii–endemic region (13,14). Although both of these reports use the previous name for C. gattii, C. neoformans serotypes B and C, their results clearly indicate that C. gattii has existed in the southeastern United States for >40 years, as opposed to the recent emergence in the PNW during the past 2 decades. A more recent report describes a C. gattii VGI isolate from an HIV-positive patient in North Carolina (15); those authors surmised that the isolate was related to travel to San Francisco, but that assumption may have to be revised.
No current molecular clock for C. gattii indicates the time required to generate the level of diversity seen in the VGI isolates. However, our findings lend credence to the hypothesis that C. gattii has circulated in the southeastern United States long enough to be considered endemic.
Dr. Lockhart is director of the Fungal Reference Laboratory, Mycotic Diseases Branch, Division of Foodborne, Waterborne, and Environmental Diseases, National Center for Emerging and Zoonotic Infectious Diseases, at CDC. His research interests include antifungal resistance and fungal population structure.
Acknowledgments
We thank Joyce Peterson, Colleen Lysen, and Carol Bolden at CDC isolate processing and molecular analysis; John Gillece, James Schupp, and Elizabeth Driebe at T-Gen for molecular analysis and discussions; Emilio DeBess of the Oregon Health Authority for providing unrelated VGI isolates; and Betsy Sellers and Carlos Franco-Paredes at Phoebe Putney Memorial Hospital, Albany, Georgia, for collecting isolates and helping identify this clade.
The work here was funded in part by research grant R21AI098059 to D.M.E.
All sequences have been deposited with the National Center for Biotechnology Information under BioProject #PRJNA295092.
References
- Diaz MR, Boekhout T, Theelen B, Fell JW. Molecular sequence analyses of the intergenic spacer (IGS) associated with rDNA of the two varieties of the pathogenic yeast, Cryptococcus neoformans. Syst Appl Microbiol. 2000;23:535–45 . DOIPubMedGoogle Scholar
- Kwon-Chung KJ, Boekhout T, Fell JW, Diaz M. Proposal to conserve the name Cryptococcus gattii against C. hondurianus and C. bacillisporus (Basidiomycota, Hymenomycetes, Tremellomycetidae). Taxon. 2002;51:804–6. DOIGoogle Scholar
- Hagen F, Khayhan K, Theelen B, Kolecka A, Polacheck I, Sionov E, Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol. 2015;78:16–48. DOIPubMedGoogle Scholar
- Wilson DE, Bennett JE, Bailey JW. Serologic grouping of Cryptococcus neoformans. Proc Soc Exp Biol Med. 1968;127:820–3. DOIPubMedGoogle Scholar
- Bennett JE, Kwon-Chung KJ, Howard DH. Epidemiologic differences among serotypes of Cryptococcus neoformans. Am J Epidemiol. 1977;105:582–6 .PubMedGoogle Scholar
- Engelthaler DM, Hicks ND, Gillece JD, Roe CC, Schupp JM, Driebe EM, Cryptococcus gattii in North American Pacific Northwest: whole-population genome analysis provides insights into species evolution and dispersal. mBiol. 2014;5:e01464–14. PubMedGoogle Scholar
- Harris JR, Lockhart SR, Sondermeyer G, Vugia DJ, Crist MB, D’Angelo MT, Cryptococcus gattii infections in multiple states outside the US Pacific Northwest. Emerg Infect Dis. 2013;19:1620–6 . DOIPubMedGoogle Scholar
- Lockhart SR, Iqbal N, Harris JR, Grossman NT, DeBess E, Wohrle R, Cryptococcus gattii in the United States: genotypic diversity of human and veterinary isolates. PLoS ONE. 2013;8:e74737. DOIPubMedGoogle Scholar
- Meyer W, Aanensen DM, Boekhout T, Cogliati M, Diaz MR, Esposto MC, Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med Mycol. 2009;47:561–70. DOIPubMedGoogle Scholar
- Gillece JD, Schupp JM, Balajee SA, Harris J, Pearson T, Yan Y, Whole genome sequence analysis of Cryptococcus gattii from the Pacific Northwest reveals unexpected diversity. PLoS ONE. 2011;6:e28550. DOIPubMedGoogle Scholar
- Sheppe WM. Torula infection in man. Am J Med Sci. 1924;167:91–107.
- Hagen F, Colom MF, Swinne D, Tintelnot K, Iatta R, Montagna MT, Autochthonous and dormant Cryptococcus gattii infections in Europe. Emerg Infect Dis. 2012;18:1618–24. DOIPubMedGoogle Scholar
- Denton JF, Di Salvo AF. The prevalence of Cryptococcus neoformans in various natural habitats. Sabouraudia. 1968;6:213–7. DOIPubMedGoogle Scholar
- Fromtling RA, Shadomy S, Shadomy HJ, Dismukes WE. Serotype B/C Cryptococcus neoformans isolated from patients in nonendemic areas. J Clin Microbiol. 1982;16:408–10 .PubMedGoogle Scholar
- Byrnes EJ III, Li W, Lewit Y, Perfect JR, Carter DA, Cox GM, First reported case of Cryptococcus gattii in the Southeastern USA: implications for travel-associated acquisition of an emerging pathogen. PLoS ONE. 2009;4:e5851. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 22, Number 6—June 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Shawn R. Lockhart, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop G11, Atlanta, GA 30329-4027, USA
Top