Volume 22, Number 7—July 2016
Dispatch
Outbreak of Vibrio parahaemolyticus Sequence Type 120, Peru, 2009
Cite This Article
Citation for Media
Abstract
In 2009, an outbreak of Vibrio parahaemolyticus occurred in Piura, Cajamarca, Lambayeque, and Lima, Peru. Whole-genome sequencing of clinical and environmental samples from the outbreak revealed a new V. parahaemolyticus clone. All the isolates identified belonged to a single clonal complex described exclusively in Asia before its emergence in Peru.
Vibrio parahaemolyticus is a marine bacterium considered to be one of the major causes of bacterial foodborne outbreaks. Infections caused by V. parahaemolyticus have shown a steady expansion in recent years, with a growing number of cases detected worldwide (1–7).
The epidemiology of V. parahaemolyticus infections in Peru has traditionally been dominated by a characteristic pattern of an increase number of cases during the summer months, corresponding to higher coastal water temperatures (8). This seasonality in the epidemic dynamics of V. parahaemolyticus infections was only altered during the emergence of cases associated with 2 major outbreaks of illnesses reported in the country, which were caused by the arrival of novel genetic variants coming from Asia (9,10). V. parahaemolyticus infections in Peru had been predominantly associated with the O4:K8 serotype and sequence type (ST) 88 until 1995 (11), when a novel genetic variant of O4:K8 emerged in the country. Infections caused by this novel variant (ST-189a) quickly spread throughout the country, replacing those caused by the ST-88 variant (10). ST-189a was replaced in 1997 as the dominant ST by the arrival of a new variant, the pandemic clone ST-3, which also originated in Asia (8,12). Infections were mostly associated with the pandemic clone throughout 1997 and 1998 and then with a less clear pattern of dominance afterwards because of the presence of multiple serotypes.
A new and large V. parahaemolyticus outbreak was detected in Peru during the austral summer of 2009. During February–March 2009, a total of 30 isolates were obtained from clinical samples of patients with symptoms of gastroenteritis. Initially illnesses were reported only in the northern cities of Peru (Cajamarca, Chiclayo, and Piura), but subsequently the outbreak extended to Lima.
Thirty V. parahaemolyticus strains isolated from this outbreak were initially investigated for the presence of virulence-related genes, serotyped, and subtyped by using pulse-field gel electrophoresis (PFGE). All strains belonged to serotype O3:K59, a serotype not previously identified in Peru; moreover, all were tdh-positive, trh-negative, and carried genes for the α variant of the type-3 secretion system 2 (T3SS2α). PFGE analysis showed that all the clinical strains shared an indistinguishable PFGE pattern (Technical Appendix Figure 1).
Environmental strains of V. parahaemolyticus isolated from shellfish collected at the central market in Lima over the course of the outbreak were also investigated. These strains (n = 4) were tdh-positive, trh-negative, T3SS2α-positive, and indistinguishable by PFGE analysis from the outbreak strains.
The genomes of 20 of those strains (18 clinical and 2 environmental) were sequenced by MiSeq (Illumina, San Diego, CA, USA) with 500 (2 × 250) cycles, 2× pair-end library with a minimum coverage of 40–120×; testing was carried out at the US Food and Drug Administration’s Center for Food Safety and Nutrition (College Park, MD, USA). Libraries were prepared with the Nextera XT DNA sample preparation kit (Illumina), according to the manufacturer’s instructions. Whole-genome sequence contigs for each strain were de novo assembled by using CLC Genomics Workbench version 7.5.1 (QIAGEN, Valencia, CA, USA).
Figure
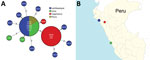
Figure. V. parahaemolyticus outbreaks in Peru, 2009. A) Minimum spanning tree showing the loci differences among Peruvian sequence type (ST) 120 strains. Ridom SeqSphere+ version 3.0.0 (http://www.ridom.de/seqsphere) identified 4,265 loci shared...
In silico multilocus sequence typing (MLST) by eBURST (13) identified all strains as belonging to a single sequence type profile, ST-120, which is the ancestral founder of clonal complex (CC) 120 (Technical Appendix Figure 2). All strains deposited in the V. parahaemolyticus MLST database belonging to CC120 originated from China. Whole-genome MLST analysis (wgMLST) using Ridom SeqSphere+ version 3.0.0 (http://www.ridom.de/seqsphere) identified 4,265 genes shared among all ST-120 strains from Peru. The genome of strain RIMD 2210633 (14) was used as reference. Ridom SeqSphere+ does a gene-by-gene mapping of the shotgun genomes against the reference genome, identifies the core genes present in all genomes, identifies variants at sequence level (single-nucleotide polymorphisms [SNPs]), and assigns alleles to each unique individual gene sequence. SNPs identified in each allele for each locus were extracted and saved into a SNP matrix to be used for further analysis. Then, Nei’s DNA distance method (15) was used for calculating the genetic distance matrix by taking the number of same/different alleles scored for each loci in each genome. In some cases, values are not found in certain loci because that gene was either missing or truncated because of its position at either end of the de novo assembled contigs. With these genetic distances, we then built either a neighbor-joining tree or minimum-spanning tree. Among those 4,265 core genes, only 20 were different from the rest. A minimum-spanning tree of these strains showed genetic uniformity among all the outbreak strains, grouping all genomes within a single complex with a central group of 6 strains (Figure). These 6 strains were indistinguishable, and the remaining strains showed minor differences ranging from 1 to 3 alleles and from 1 to 5 SNPs (Technical Appendix Figure 3). Furthermore, environmental strains showed identical allelic profiles and sequences to the outbreak strains, which represent evidence supporting the domestic source of the seafood originating the infections. The shellfish predominantly comprised bivalve mollusk species collected from warm areas of the north of the country where the outbreak originated and that are shipped daily to the central market in Lima.
A wgMLST analysis of the outbreak isolates with 236 V. parahaemolyticus genomes available in GenBank grouped the ST-120 isolates from Peru in a single cluster that exclusively included isolates from China (Technical Appendix Figure 4). This finding constitutes additional support to the findings observed with the use of the available MLST data (Technical Appendix Figure 2), which show genetically similar strains in very distant locations. By wgMLST analysis, ST-120 isolates from Peru differed from 2 isolates isolated in China in 1992 (S016) and 1993 (S018) by 48 and 259 alleles, respectively. The fact that these 2 strains were isolated during the 1990s might explain why they are so different from the Peru ST-120 strains. Expanding the analysis to other genomes of ST-120 recently isolated from China or Southeast Asia might identify more closely related strains.
Taken together, our findings reveal another example of the emergence of an Asian variant of V. parahaemolyticus in Peru associated with seafood consumption. The arrival of ST-120 strains in Peru represents a third instance of an introduction of Asian populations of pathogenic V. parahaemolyticus to the Pacific coasts of South America, and, together with the arrival of strains of the seventh pandemic of cholera in 1991, substantiates the existence of recurrent flux of pathogenic Vibrio populations between both sides of the Pacific Ocean. Asian and Peruvian coasts are intermittently interconnected through the movement of water displaced by El Niño episodes. These 4 introduction events of pathogenic Vibrio strains in Peru occurred just before the arrival of tropical El Niño waters to the Peruvian coasts, which suggests that the introduction of foreign populations of Vibrio could be mediated by El Niño events, as previously suggested (8).
In conclusion, this study stresses the importance of the application of genomic epidemiology for the routine investigation of outbreaks and surveillance as an efficient and high-resolution tool for tracing the dissemination of pathogens and diseases on a global scale. This latter information is critical to detect the emergence of novel genetic variants, understand the colonization history of pathogens, and assess potential sources and scenarios contributing to the emergence of disease.
Dr. Gonzalez-Escalona is a research microbiologist at the Center for Food Safety and Applied Nutrition of the US Food and Drug Administration, College Park, Maryland, USA. His research focusses on food safety, molecular identification and characterization of foodborne pathogens, and the use of whole-genome sequencing for subtyping and source tracking.
Acknowledgments
The authors thank the epidemiologists and public health offices from the Instituto Nacional de Salud and the regional laboratories in Peru for providing the data and isolates, and Elizabeth Reed for her editorial assistance on this manuscript.
This project was supported by the University of Bath project VB-BB1JMU and the US Food and Drug Administration’s Foods Program Intramural Funds.
References
- Turner JW, Paranjpye RN, Landis ED, Biryukov SV, González-Escalona N, Nilsson WB, Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS One. 2013;8:e55726. DOIPubMedGoogle Scholar
- Haendiges J, Timme R, Allard MW, Myers RA, Brown EW, Gonzalez-Escalona N. Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012–2013) and comparisons to a locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front Microbiol. 2015;6:125 .DOIPubMedGoogle Scholar
- Banerjee SK, Kearney AK, Nadon CA, Peterson CL, Tyler K, Bakouche L, Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J Clin Microbiol. 2014;52:1081–8. DOIPubMedGoogle Scholar
- Theethakaew C, Feil EJ, Castillo-Ramírez S, Aanensen DM, Suthienkul O, Neil DM, Genetic relationships of Vibrio parahaemolyticus isolates from clinical, human carrier, and environmental sources in Thailand, determined by multilocus sequence analysis. Appl Environ Microbiol. 2013;79:2358–70. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Baker-Austin C, Jones JL, Newton AE, Gonzalez-Aviles GD, DePaola A. Spread of Pacific Northwest Vibrio parahaemolyticus strain. N Engl J Med. 2013;369:1573–4. DOIPubMedGoogle Scholar
- Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin Microbiol Rev. 2007;20:39–48. DOIPubMedGoogle Scholar
- Haendiges J, Rock M, Myers RA, Brown EW, Evans P, Gonzalez-Escalona N. Pandemic Vibrio parahaemolyticus, Maryland, USA, 2012. Emerg Infect Dis. 2014;20:718–20. DOIPubMedGoogle Scholar
- Martinez-Urtaza J, Huapaya B, Gavilan RG, Blanco-Abad V, Ansede-Bermejo J, Cadarso-Suarez C, Emergence of Asiatic Vibrio diseases in South America in phase with El Niño. Epidemiology. 2008;19:829–37. DOIPubMedGoogle Scholar
- Gil AI, Miranda H, Lanata CF, Prada A, Hall ER, Barreno CM, O3:K6 serotype of Vibrio parahaemolyticus identical to the global pandemic clone associated with diarrhea in Peru. Int J Infect Dis. 2007;11:324–8 .DOIPubMedGoogle Scholar
- González-Escalona N, Gavilan RG, Brown EW, Martinez-Urtaza J. Transoceanic spreading of pathogenic strains of Vibrio parahaemolyticus with distinctive genetic signatures in the recA gene. PLoS One. 2015;10:e0117485. DOIPubMedGoogle Scholar
- Gavilan RG, Zamudio ML, Martinez-Urtaza J. Molecular epidemiology and genetic variation of pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl Trop Dis. 2013;7:e2210. DOIPubMedGoogle Scholar
- González-Escalona N, Gavilan RG, Brown EW, Martinez-Urtaza J. Transoceanic spreading of pathogenic strains of Vibrio parahaemolyticus with distinctive genetic signatures in the recA gene. PLoS One. 2015;10:e0117485. DOIPubMedGoogle Scholar
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–30. DOIPubMedGoogle Scholar
- Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet. 2003;361:743–9. DOIPubMedGoogle Scholar
- Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol. 1983;19:153–70. DOIPubMedGoogle Scholar
Figure
Cite This ArticleTable of Contents – Volume 22, Number 7—July 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Address for correspondence. Jaime Martinez-Urtaza, Milner Centre for Evolution, Department of Biology and Biochemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
Top