Volume 31, Supplement—May 2025
SUPPLEMENT ISSUE
Supplement
Genomic Characterization of Escherichia coli O157:H7 Associated with Multiple Sources, United States
Cite This Article
Citation for Media
Abstract
In the United States, Shiga toxin–producing Escherichia coli (STEC) outbreaks cause >265,000 infections and cost $280 million annually. We investigated REPEXH01, a persistent strain of STEC O157:H7 associated with multiple sources, including romaine lettuce and recreational water, that has caused multiple outbreaks since emerging in late 2015. By comparing the genomes of 729 REPEXH01 isolates with those of 2,027 other STEC O157:H7 isolates, we identified a highly conserved, single base pair deletion in espW that was strongly linked to REPEXH01 membership. The biological consequence of that deletion remains unclear; further studies are needed to elucidate its role in REPEXH01. Additional analyses revealed that REPEXH01 isolates belonged to Manning clade 8; possessed the toxins stx2a, stx2c, or both; were predicted to be resistant to several antimicrobial compounds; and possessed a diverse set of plasmids. Those factors underscore the need to continue monitoring REPEXH01 and clarify aspects contributing to its emergence and persistence.
Shiga toxin–producing Escherichia coli (STEC) outbreaks associated with produce were first identified in 1991, and the trend of produce-associated STEC outbreaks remains prevalent, among which romaine lettuce is the most common leafy green outbreak vehicle (1–4). Each year in the United States, >265,000 STEC infections occur, costing $280 million and resulting in ≈3,600 hospitalizations and ≈30 deaths (4,5). E. coli O157:H7, a specific serotype of STEC, causes ≈25% of those infections and ≈67% of all STEC deaths (5). STEC O157:H7 infections often induce abdominal cramps, vomiting, and bloody diarrhea. In particularly severe cases, a rare condition known as hemolytic uremic syndrome (HUS) develops, which can cause anemia, acute renal failure, and death (6). STEC O157:H7 outbreaks are commonly linked to consumption of leafy greens or beef. Although nearly 60% of STEC O157:H7 infections have been attributed to vegetable row crops, a category that includes leafy greens, ruminants, especially cattle, are the suspected primary STEC O157:H7 reservoir (7–9). During 2009–2018, 32 STEC O157:H7 outbreaks in the United States and Canada were linked to contaminated leafy greens (4).
Since April 2017, nine separate outbreaks of the same strain of STEC O157:H7, hereafter referred to as REPEXH01, have occurred (Table 1). A large REPEXH01 outbreak affecting 37 states occurred in 2018, from which 238 STEC O157:H7 infections, 104 hospitalizations, 28 cases of HUS, and 5 deaths were reported (3). Most (85%) interviewed patients reported consuming romaine lettuce, and a subsequent investigation linked those infections to romaine lettuce grown in the Yuma, Arizona, region of the United States (3). By March 29, 2024, the United States reported 762 persons in 46 states infected with the REPEXH01 strain, and new infections continue to be identified. In this study, we compared whole-genome sequences of 729 REPEXH01 isolates with 2,027 other STEC O157:H7 isolates to examine genomic factors in REPEXH01 that might have contributed to the emergence and public health impacts of that strain.
Sequence Selection and Retrieval
We used sequences from 729 REPEXH01 isolates and 598 closely related isolates previously classified as REPEXH01 for this study. All isolates were in PulseNet (https://www.cdc.gov/pulsenet/index.html) and had whole-genome sequences available in the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov) (Appendix 1). To compare a diverse collection of STEC O157:H7, we randomly selected 1,429 non-REPEXH01 STEC O157:H7 isolates, for a total of 2,756 genomes analyzed. That total accounts for roughly 20% of all 13,778 STEC O157:H7 isolates within PulseNet that had whole-genome sequences available in NCBI as of September 5, 2023. We downloaded whole-genome sequences from GenBank and assemblies and raw reads from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) during May 23–August 1, 2023 (Appendix 2 Table 1). We used Genbank annotated genomes when available and used Prokka version 1.14.5 (10) to annotate SRA genomes that did not have annotations.
Identification of Genomic Features
We used Roary version 3.11.2 (11) to perform pangenome analysis on Prokka-annotated genomes, then screened pangenomes for linkage to REPEXH01 isolates by using Scoary version 1.6.16 (12). Because those steps are computationally intensive, we used a subset of genomes comprising 181 current and 103 former REPEXH01 isolates and 2 closely related non-REPEXH01 isolates. We identified multiple alleles of espW, a known virulence gene, in that initial dataset and subsequently profiled the expanded dataset (n = 2,756) for those alleles and their association with REPEHX01 (13,14) (Appendix 2). We screened assemblies for antimicrobial resistance determinants, plasmid determinants, antimicrobial resistance determinant–associated point mutations, membership in O157 clades (hereafter referred to as Manning clades), and stx subtypes (Appendix 1).
Phylogenetic Reconstruction
From the subset of genomes profiled for pangenome analysis, we constructed a single-nucleotide polymorphism (SNP) analysis by using Lyve-SET version 1.1.4f (https://github.com/lskatz/lyve-SET) (15) and presets for Escherichia using the single chromosomal contig of 2018C-3602 (BioSample accession no. SAMN08964444) as the reference. We used Gubbins version 3.0.0 (Sanger, https://sanger-pathogens.github.io/gubbins) to generate a recombination-free SNP alignment from the Lyve-SET core alignment (15,16). We then generated a time-scaled phylogenetic tree from the SNP alignment for a subset of 286 isolates in BEAST2 version 2.6.3 (17), accounting for constant sites and using bModelTest version 1.2.1 (18) to average across appropriate substitution models. We used BioNumerics version 7.6.3 (Applied Maths, http://www.applied-maths.com) to construct an allele-based dendrogram for 2,754 isolates by using UPGMA as the clustering technique. We excluded 2 isolates from the dendrogram because the submitting state agencies had requested those isolates be removed from PulseNet.
Prophage Detection
We detected prophage sequences in the reference genome and categorized their genes by using the PHASTER online phage search tool (19,20). We used BLASTn version 2.14.0 (https://blast.ncbi.nlm.nih.gov) to search all espW-containing contigs for prophages (Appendix 1).
Obtaining and Visualizing Isolate Metadata
Unless otherwise specified, we obtained all metadata associated with isolates in this study from the System for Enteric Disease Response, Investigation, and Coordination (SEDRIC) (https://www.cdc.gov/foodsafety/outbreaks/tools/sedric.html) or the PulseNet national database (21). We visualized data alongside phylogenies by using the Interactive Tree of Life version 5 webtool (https://itol.embl.de) (22).
Epidemiology of REPEXH01
All REPEXH01 isolates belonged to Manning clade 8, the clade most strongly correlated with patients developing HUS (23,24). In fact, nearly every outbreak associated with the REPEXH01 strain included cases of HUS, and an average of 11% (median 9%) of reported illnesses displayed HUS (Table 1). Of the 729 REPEXH01 isolates, all possessed stx2a, stx2c, or both: 699 (96%) isolates possessed stx2a, 574 (79%) possessed stx2c, and 544 (75%) possessed both stx2a and stx2c (Appendix 2 Table 3). Because all REPEXH01 isolates belonged to Manning clade 8, those isolates likely all possessed stx2a, and the absence of stx2a in 4% of isolates was likely an artifact of the genome assemblies (23,24).
Relationship between espW and REPEXH01
Figure 1

Figure 1. Time-calibrated tree for 286 former and current REPEXH01 isolates with associated metadata used for genomic characterization of Escherichia coliO157:H7 associated with multiple sources, United States. The tree was...
Figure 2
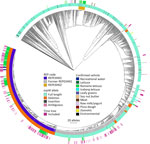
Figure 2. Allele-based core genome multilocus sequence typing dendrogram for 2,752 STEC O157:H7 isolates and associated metadata used for genomic characterization of Escherichia coliO157:H7 associated with multiple sources, United States....
We performed a preliminary Roary/Scoary pangenome analysis on a subset of 264 isolates, which indicated that the presence of espW was linked to membership in REPEXH01, but that same linkage was absent when analyzing the 286 isolates in the time-scaled tree (Figure 1). Closer inspection revealed that espW was in all isolates but often possessed a conserved single base pair deletion, and that deletion appeared to be linked to REPEXH01. We confirmed that hypothesis by analyzing the espW alleles in 2,756 isolates, 729 of which were REPEXH01, 598 were former REPEXH01 isolates, and the other 1,429 were a random sampling of all other STEC O157:H7 isolates in the PulseNet database that had publicly available genomes in NCBI (Table 2, Figure 2; Appendix 2 Table 2). We used a χ2 statistical test, ignoring ambiguous data, to examine the relationship between espW alleles and REPEXH01 membership and found the association between those variables was significant (p<0.0001). REPEXH01 isolates were more likely to have the deletion than other STEC O157:H7 isolates.
The deletion in espW consisted of the loss of a single adenine residue, converting a homopolymer within codons 174–176 from 8 adenine to 7 adenine residues. That deletion introduced a frameshift that ultimately resulted in an early termination codon. We observed insertion of an adenine residue, from 8 to 9 residues, within the same locus in 22 isolates. That insertion also introduced an early termination codon.
REPEXH01 Emergence
Analysis of 286 current and former REPEXH01 isolates revealed that the strain emerged around December 23, 2015 (95% highest posterior density interval March 5, 2015–September 4, 2016), before it was detected in clinical cases in April 2017 (Figure 1). That phylogeny appeared to suggest that members of REPEXH01 shared a common ancestor and the single base pair deletion in espW associated with REPEXH01 appeared to coincide with the emergence of the REPEXH01 strain in late 2015 (Figure 1).
espW Association with STEC O157:H7 Prophages
Figure 3

Figure 3. Prophage architecture for most espW-containing isolates from a genomic characterization of Escherichia coliO157:H7 associated with multiple sources, United States. This locus was annotated using PROKKA version...
Examining the gene synteny surrounding espW in the reference sequence for REPEXH01 (BioSample accession no. SAMN0896444) showed that many neighboring genes appeared to be of phage origin. Analyzing that genome using PHASTER (20) revealed that espW was contained within a putative prophage that was most closely related to Escherichia phage 500465-1 (GenBank accession no. NC_049342.1) (Figure 3). We examined the genomic regions containing espW, and most isolates possessed espW within the same putative prophage (Appendix 2 Table 2). Although we detected additional loci in ≈43 isolates, most were of phage origin. Of the 2,626 isolates with assembled contigs that contained espW, 87% (n = 2,292) possessed espW in or near a putative prophage region (Appendix 2 Table 2). One isolate (SRA accession no. SRR93211959) possessed espW directly adjacent to a prophage in what appeared to be an effector exchange locus (13). Another isolate (SRA accession no. SRR6870099) contained espW in a nonprophage region. In the other 332 (13%) isolates, presence of espW in a phage-associated region was ambiguous.
Additional REPEXH01 Genomic Features
We evaluated antimicrobial resistance determinants in REPEXH01 (Table 3; Appendix 2 Table 3). REPEXH01 is known to be resistant to several antimicrobial drugs and our dataset confirmed that resistance (25). Of note, our results predicted that >99% of REPEXH01 isolates would be resistant to aminoglycosides, folate pathway inhibitors, phenicols, quaternary ammonium compounds, sulfonamides, and tetracylines. However, data predict few isolates would be resistant to cephalosporins (<2%), fluoroquinolones (<1%), or penicillins (<1%).
We also investigated REPEXH01 plasmids (Table 4; Appendix 2 Table 3) and detected >1 plasmid replicons in >95% of isolates. Most isolates possessed the IncFIB replicon, IncFIA replicon, or both replicons, but other replicons were not as prevalent. Approximately 9% of isolates contained IncFII replicons, but IncI1-Iγ, IncI2, IncB/O/K/Z, Col, IncX4, and pEC4115 were detected in <5% of isolates.
A key finding in this study was identification of a SNP mutation in the espW gene that is largely characteristic of the REPEXH01 strain. The EspW protein has been shown to be secreted by a type III secretion system (T3SS) in E. coli O157:H7 and was previously observed within effector exchange locus (13). Once secreted into the host intestinal epithelial cell, EspW reorganizes host-cell actin in a Rac1-dependent manner to enable extracellular attachment (14). A Pseudomonas syringae homolog of that protein, HopW1, has been shown to solubilize cytosolic actin when injected into plant cells by a T3SS, which disrupts normal localization of proteins and might interfere with the plant immune response (26). The T3SS and secretory proteins such as EspA have been shown to play integral roles in the colonization of the surface of leaves and deeper tissues of the phyllosphere in spinach and lettuce, where STEC O157:H7 can continue to grow under favorable conditions (27,28).
However, the biological significance of the single base pair deletion in espW remains unclear. That deletion could be an example of a gene truncation; another study observed truncations of espW in other pathogenic strains of E. coli (14). Alternatively, the resulting frame shift might silence expression of espW, or espW might be regulated by a homopolymeric tract mechanism where slippage of RNA polymerase could produce heterogenous transcripts, some of which could encode the in-frame functional gene product (29). In each of those scenarios, reduced EspW could promote colonization of romaine lettuce through several mechanisms. For example, EspW might elicit an immune response from an infected plant, causing stomata to close, thus restricting access to the interior of leaves by colonizing STEC. Alternatively, EspW could function like HopW1 and cause a more severe infection in plant tissues, lowering the likelihood that the infected leaves are harvested and consumed. Further experiments are required to elucidate the role of that single base pair deletion in REPEXH01 isolates.
In this study, we performed key molecular profiling to provide information on molecular attributes of REPEXH01. Certain stx subtypes are associated with more severe disease, and the prevalence of stx2a in REPEXH01 highlights the need for surveillance of this strain (30). All isolates of this strain belonged to Manning clade 8, the clade most strongly correlated with poor disease outcomes (23,24). Nearly all REPEXH01 isolates possessed antimicrobial resistance determinants, but that finding does not have direct clinical significance because antimicrobial drugs are not indicated for treating STEC infections because those drugs can increase toxin concentrations in the patient (25). However, the plasmids observed in REPEXH01 isolates have been implicated in horizontal gene transfer, and those plasmids were in >95% of REPEXH01 isolates (Appendix 2 Table 3) (31). Taken together, those findings suggests that although the presence of antimicrobial resistance determinants has minimal effects on clinical outcomes of STEC infections, and REPEXH01 isolates could still serve as a reservoir of antimicrobial resistance.
Among the limitations of this study, although we included all current and former REPEXH01 isolates in this study, we only screened 20% of the total STEC O157:H7 isolates to decrease the computational demand of the analyses. That subsampling has the potential to bias the data, but the random selection of non-REPEXH01 STEC O157:H7 genomes might alleviate that bias. The genomes used in this study were primarily derived from short-read sequencing technology, and most were at the draft level, indicating that the replicons had not been fully assembled. Although use of draft genomes could result in espW being erroneously called absent, steps such as read recruitment using ARIBA (https://github.com/sanger-pathogens/ariba) helped mitigate those potential errors.
REPEXHO1 is a persistent strain of STEC O157:H7 that we estimate emerged in late 2015, before the detection of clinical cases beginning in April 2017. We detected a single base pair deletion in the espW virulence gene in >99% of REPEXH01 isolates but in only a few (<4%) non-REPEXH01 STEC O157:H7 isolates (Table 2). That deletion can be useful as a genomic signature of this strain for molecular surveillance and as a subject of future research to clarify the strain’s evolution. Additional research addressing the role of the single base pair mutation in this strain’s colonization and survival on leafy vegetables could yield valuable insights.
In summary, REPEHX01 belongs to E. coli O157:H7 Manning Clade 8, and most isolates possess stx2a, both factors that are associated with severe clinical outcomes. Those factors, along with its harboring of multiple resistance determinants, underscore the continued need to monitor REPEXH01 and understand factors contributing to its emergence and persistence.
Dr. Wirth is bioinformatician on the Molecular Virology Team in the Viral Vaccine-Preventable Disease Branch, Division of Viral Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA. His research interests include the application of computational techniques to microbiological problems, especially those involving the evolution and physiology of human pathogens.
Acknowledgments
We thank Kaitlin Tagg for providing subject matter expertise on plasmid classification. We thank both Kaitlin Tagg and Hattie Webb for their helpful discussions and insightful comments.
This work was made possible by support from the Office of Advanced Molecular Detection at the Centers for Disease Control and Prevention and is covered by activities approved by the Centers for Disease Control and Prevention Institutional Review Board (approval no. 7172).
References
- Rangel JM, Sparling PH, Crowe C, Griffin PM, Swerdlow DL. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982-2002. Emerg Infect Dis. 2005;11:603–9. DOIPubMedGoogle Scholar
- Dewey-Mattia D, Manikonda K, Hall AJ, Wise ME, Crowe SJ. Surveillance for foodborne disease outbreaks—United States, 2009–2015. MMWR Surveill Summ. 2018;67:1–11. DOIPubMedGoogle Scholar
- Bottichio L, Keaton A, Thomas D, Fulton T, Tiffany A, Frick A, et al. Shiga toxin–producing Escherichia coli infections associated with romaine lettuce—United States, 2018. Clin Infect Dis. 2020;71:e323–30. DOIPubMedGoogle Scholar
- Marshall KE, Hexemer A, Seelman SL, Fatica MK, Blessington T, Hajmeer M, et al. Lessons learned from a decade of investigations of Shiga toxin–producing Escherichia coli outbreaks linked to leafy greens, United States and Canada. Emerg Infect Dis. 2020;26:2319–28. DOIPubMedGoogle Scholar
- Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, et al. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis. 2011;17:7–15. DOIPubMedGoogle Scholar
- Interagency Food Safety Analytics Collaboration. Foodborne illness source attribution estimates for 2020 for Salmonella, Escherichia coli O157, and Listeria monocytogenes using multi-year outbreak surveillance data, United States. US Department of Health and Human Services, Centers for Disease Control and Prevention, Food and Drug Administration, US Department of Agriculture Food Safety and Inspection Service, editors. Atlanta and Washington; The Departments; 2020.
- Chen JC, Patel K, Smith PA, Vidyaprakash E, Snyder C, Tagg KA, et al. Reocurring Escherichia coli O157:H7 strain linked to leafy greens–associated outbreaks, 2016–2019. Emerg Infect Dis. 2023;29:1895–9. DOIPubMedGoogle Scholar
- Bielaszewska M, Schmidt H, Liesegang A, Prager R, Rabsch W, Tschäpe H, et al. Cattle can be a reservoir of sorbitol-fermenting shiga toxin-producing Escherichia coli O157:H(-) strains and a source of human diseases. J Clin Microbiol. 2000;38:3470–3. DOIPubMedGoogle Scholar
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. DOIPubMedGoogle Scholar
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3. DOIPubMedGoogle Scholar
- Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17:1. DOIGoogle Scholar
- Tobe T, Beatson SA, Taniguchi H, Abe H, Bailey CM, Fivian A, et al. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. P Proc Natl Acad Sci U S A. 2006;103:14941–6.
- Sandu P, Crepin VF, Drechsler H, McAinsh AD, Frankel G, Berger CN. The enterohemorrhagic Escherichia coli effector EspW triggers actin remodeling in a Rac1-dependent manner. Infect Immun. 2017;85:e00244–17. DOIPubMedGoogle Scholar
- Katz LS, Griswold T, Williams-Newkirk AJ, Wagner D, Petkau A, Sieffert C, et al. A comparative analysis of the lyve-SET phylogenomics pipeline for genomic epidemiology of foodborne pathogens. Front Microbiol. 2017;8:375. DOIPubMedGoogle Scholar
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:
e15 . DOIPubMedGoogle Scholar - Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLOS Comput Biol. 2014;10:
e1003537 . DOIPubMedGoogle Scholar - Bouckaert RR, Drummond AJ. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol Biol. 2017;17:42. DOIPubMedGoogle Scholar
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39):W347-52.
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44(W1):
W16-21 . DOIPubMedGoogle Scholar - Swaminathan B, Barrett TJ, Hunter SB, Tauxe RV; CDC PulseNet Task Force. PulseNet: the molecular subtyping network for foodborne bacterial disease surveillance, United States. Emerg Infect Dis. 2001;7:382–9. DOIPubMedGoogle Scholar
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6. DOIPubMedGoogle Scholar
- Manning SD, Motiwala AS, Springman AC, Qi W, Lacher DW, Ouellette LM, et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc Natl Acad Sci U S A. 2008;105:4868–73. DOIPubMedGoogle Scholar
- Iyoda S, Manning SD, Seto K, Kimata K, Isobe J, Etoh Y, et al. Phylogenetic clades 6 and 8 of enterohemorrhagic Escherichia coli O157:H7 with particular stx subtypes are more frequently found in isolates from hemolytic uremic syndrome patients than from asymptomatic carriers. Open Forum Infect Dis. 2014;1:
ofu061 . DOIPubMedGoogle Scholar - Centers for Disease Control and Prevention. Persistent strain of E. coli O157:H7 (REPEXH01) linked to multiple sources [cited 2024 Mar 7]. https://www.cdc.gov/ecoli/php/data-research/repexh01-e-coli-o157h7.html
- Kang Y, Jelenska J, Cecchini NM, Li Y, Lee MW, Kovar DR, et al. HopW1 from Pseudomonas syringae disrupts the actin cytoskeleton to promote virulence in Arabidopsis. PLoS Pathog. 2014;10:
e1004232 . DOIPubMedGoogle Scholar - Xicohtencatl-Cortes J, Sánchez Chacón E, Saldaña Z, Freer E, Girón JA. Interaction of Escherichia coli O157:H7 with leafy green produce. J Food Prot. 2009;72:1531–7. DOIPubMedGoogle Scholar
- Saldaña Z, Sánchez E, Xicohtencatl-Cortes J, Puente JL, Girón JA. Surface structures involved in plant stomata and leaf colonization by shiga-toxigenic Escherichia coli o157:h7. Front Microbiol. 2011;2:119. DOIPubMedGoogle Scholar
- Orsi RH, Bowen BM, Wiedmann M. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genomics. 2010;11:102. DOIPubMedGoogle Scholar
- Byrne L, Adams N, Jenkins C. Association between Shiga toxin-producing Escherichia coli O157:H7 stx gene subtype and disease severity, England, 2009–2019. Emerg Infect Dis. 2020;26:2394–400. DOIPubMedGoogle Scholar
- Redondo-Salvo S, Fernández-López R, Ruiz R, Vielva L, de Toro M, Rocha EPC, et al. Pathways for horizontal gene transfer in bacteria revealed by a global map of their plasmids. Nat Commun. 2020;11:3602. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: May 06, 2025
Table of Contents – Volume 31, Supplement—May 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Joseph S. Wirth, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop H18-5, Atlanta, GA 30329-4018, USA
Top