Volume 22, Number 7—July 2016
Letter
Pegivirus Infection in Domestic Pigs, Germany
Cite This Article
Citation for Media
To the Editor: The family Flaviviridae includes many human and animal virus pathogens. Recently, in addition to the genera Flavivirus, Hepacivirus, and Pestivirus, a fourth genus, Pegivirus, has been identified (1). In addition to human pegiviruses, a range of phylogenetic, highly divergent pegiviral sequences have been identified in various animal species, including primates, bats, rodents, and horses (2). We report the detection of a porcine pegivirus (PPgV) in serum samples from pigs.
Initially, we investigated pooled serum samples by using high-throughput sequencing methods and isolated RNA from individual porcine serum samples by using the QIAmp Viral RNA Mini Kit (QIAGEN, Hilden, Germany). We prepared libraries compatible with Illumina (San Diego, CA, USA) sequencing from pooled samples and individual serum samples by using the ScriptSeq version 2 RNA-Seq Library Preparation Kit (Epicenter, Madison, WI, USA) and sequenced them by using a HiSeq 2500 (2 × 150 cycles paired-end; Illumina) for pooled samples and MiSeq (2 × 250 cycles paired-end; Illumina) for individual samples (3).
We conducted quantitative reverse transcription PCR (RT-PCR) by using a Quantitect-SYBR Green Assay (QIAGEN) and primers PPgV_fwd: 5′-CTGTCTATGCTGGTCACGGA-3′ and PPgV_rev: 5′-GCCATAGAACGGGAAGTCGC-3′. By using high-throughput sequencing of the pooled serum sample library (23,167,090 reads), we identified 1 contig (4,582 bp) that had distant nucleotide sequence simi-larity to bat pegivirus (69% and 4% sequence coverage) and 2 contigs (2,683 bp and 665 bp) that had 73% sequence coverage, thereby covering 8% and 37% of the identified sequence. RT-PCR with primers designed on basis of recovered sequences identified the sample containing pegivirus sequences. Subsequent MiSeq analysis (7,085,595 reads) of an RNA library prepared from a sample from 1 animal identified 1 contig (9,145 nt) with sequence similarity to pe-givirus sequences.
We performed 3′ end completion of the viral genome by rapid amplification of cDNA ends and identified the entire open reading frame of PPgV_903 encoding 2,972 aa (GenBank accession no. KU351669). Analysis of the pegivirus 5′-untranslated region identified a highly structured internal ribosome entry site motif (Technical Appendix), which was similar in structure to previously described 5′ untranslated region structures of other pegiviruses (4,5).
Pegiviruses do not encode a protein homologous to the capsid protein of other viruses of the family Flaviviridae, another common feature of pegiviruses (6). The presence of cleavage sites for cellular signal peptidases and viral proteases indicates that, similar to polyproteins of other pegiviruses and members of the genus Hepacivirus, the pegivirus polyprotein NH2-E1- E2-Px-NS2-NS3-NS4A-NS4B-NS5A-NS5B-COOH (E [envelope], NS [nonstructural], and Px [protein X]) is cleaved co-translationally and posttranslationally.
We tested 3 additional animals from the same breeding cohort for virus RNA at irregular intervals for 22 months. One animal was positive for pegivirus RNA for 7 months, and the other 2 animals had pegivirus RNA in serum for 16 and 22 months. None of these animals showed obvious clinical signs attributable to virus infection. Follow-up investigation of 455 serum samples from 37 swine holdings from Germany identified 10 (2.2%) samples from 6 pig holdings that contained pegivirus RNA. We obtained 2 additional near full-length genomic sequences (PPgV_80F and PPgV_S8-7) from 2 animals in different herds by high-throughput sequencing, RT-PCR, and Sanger sequencing (GenBank accession nos. KU351670 and KU351671).
Figure
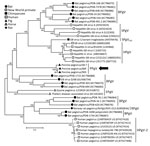
Figure. Phylogenetic analysis of human and animal pegiviruses. We constructed a maximum-likelihood tree on the basis of the complete coding region and used the general time reversible model for modeling of substitutions....
Phylogenetic analyses of complete coding regions showed the close relationship of the 3 pegivirus sequences from Germany. These 3 sequences formed a separate clade within the genus Pegivirus (Figure). Pairwise comparison between PPgV_903 and the other 2 pegivirus sequences showed strong nucleotide identities (96.0%–98.4%). A distance scan over the entire polyprotein showed genetic distance to other pegiviruses and demonstrated that NS3 and NS5B contain the most conserved regions among pegivirus polyproteins (Technical Appendix).
In horses, 2 distinct pegiviruses that had different potentials to cause clinical disease in infected animals have been described (4,7). No obvious clinical effects were observed in pegivirus-infected animals during our study. However, potential consequences of viral infection for animal health and food production need to be explored more closely under field and experimental conditions. Pegiviruses can interact with the immune system of the host. Co-infection with human pegivirus and HIV can have beneficial effects, which result in decreased retroviral loads and delayed disease progression (8).
It will be useful to investigate whether co-infections with pegiviruses can influence clinical manifestations of infectious diseases of swine, including multifactorial diseases such as postweaning multisystemic wasting syndrome, in which unknown immune modulating virus infections have been suggested to influence the degree of clinical illness (9). RNA viruses have considerable potential to adapt to new environmental conditions and to overcome host restrictions (10). Until now, the host tropism of PPgV has not been investigated in detail. Therefore, additional studies will be required to elucidate whether the spectrum of potential hosts might include other farm or companion animals, and whether the virus might be able to infect humans.
Acknowledgments
We thank Jens Böttcher, Thomas Große Beilage, Diana Meemken, Alexandra von Altrock, and Cornelia Schwennen for collecting serum samples; Polina Parfentev for providing excellent technical assistance; and Daniela Indenbirken for providing support in preparation of the RNA library.
This study was supported by the German Center for Infection Research/Thematic Translational Unit Emerging Infections.
References
- Stapleton JT, Bukh J, Muerhoff AS, Foung S, Simmonds P. Assignment of human, simian and bat pegiviruses (previously described as GBV-A, GBV-C, and GBV-D) as members of a new genus (Pegivirus) within the Flaviviridae [cited 2015 Oct 21]. http://www.ictvonline.org/proposals/2012.011a-dV.A.v2.Pegivirus.pdf
- Thézé J, Lowes S, Parker J, Pybus OG. Evolutionary and phylogenetic analysis of the hepaciviruses and pegiviruses. Genome Biol Evol. 2015;7:2996–3008 .DOIPubMedGoogle Scholar
- Baechlein C, Fischer N, Grundhoff A, Alawi M, Indenbirken D, Postel A, Identification of a novel hepacivirus in domestic cattle from Germany. J Virol. 2015;89:7007–15. DOIPubMedGoogle Scholar
- Kapoor A, Simmonds P, Cullen JM, Scheel TK, Medina JL, Giannitti F, Identification of a pegivirus (GB virus-like virus) that infects horses. J Virol. 2013;87:7185–90. DOIPubMedGoogle Scholar
- Simons JN, Desai SM, Schultz DE, Lemon SM, Mushahwar IK. Translation initiation in GB viruses A and C: evidence for internal ribosome entry and implications for genome organization. J Virol. 1996;70:6126–35.PubMedGoogle Scholar
- Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol. 2011;92:233–46 .DOIPubMedGoogle Scholar
- Chandriani S, Skewes-Cox P, Zhong W, Ganem DE, Divers TJ, Blaricum AJ, Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. Proc Natl Acad Sci U S A. 2013;110:E1407–15. DOIPubMedGoogle Scholar
- Schwarze-Zander C, Blackard JT, Rockstroh JK. Role of GB virus C in modulating HIV disease. Expert Rev Anti Infect Ther. 2012;10:563–72 .DOIPubMedGoogle Scholar
- Grau-Roma L, Fraile L, Segalés J. Recent advances in the epidemiology, diagnosis and control of diseases caused by porcine circovirus type 2. Vet J. 2011;187:23–32. DOIPubMedGoogle Scholar
- Rosenberg R. Detecting the emergence of novel, zoonotic viruses pathogenic to humans. Cell Mol Life Sci. 2015;72:1115–25. DOIPubMedGoogle Scholar
Figure
Cite This Article1These authors contributed equally to this article.
Related Links
Table of Contents – Volume 22, Number 7—July 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Paul Becher, Institute of Virology, Department of Infectious Diseases, University of Veterinary Medicine, Buenteweg 17, 30559 Hannover, Germany
Top