Volume 23, Number 7—July 2017
Research Letter
Importation of Zika Virus from Vietnam to Japan, November 2016
Figure
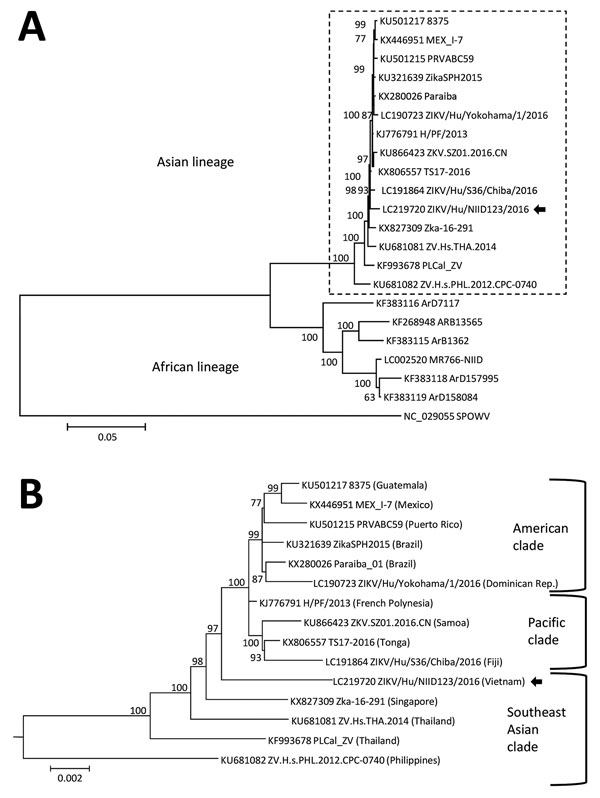
Figure. Phylogenetic analysis of the Zika virus sequence derived from a patient returning to Japan from Vietnam in November 2016. The phylogenetic tree was based on a nearly complete genome and constructed by using the maximum-likelihood method (MEGA 7.0, http://www.kumarlab.net/publications). The sequence derived from the patient is indicated with an arrow. A) The phylogenetic tree based on a nearly full-length region. B) The expanded Asian lineage branch (dotted box in panel A). Scale bars indicate nucleotide substitutions per site.
Page created: June 19, 2017
Page updated: June 19, 2017
Page reviewed: June 19, 2017
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.