Volume 24, Number 12—December 2018
CME ACTIVITY - Synopsis
Reemergence of St. Louis Encephalitis Virus in the Americas
Figure 2
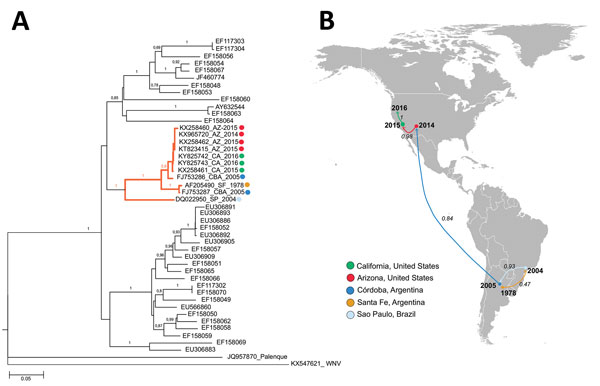
Figure 2. Phylogeny and spread of St. Louis encephalitis virus (SLEV) in the Americas. A) Multiple sequence alignment of 44 complete envelope protein SLEV sequences obtained from GenBank. The orange highlighted cluster contained the emerging SLEV strains isolated in Argentina, Brazil, and western United States. Alignment was performed by ClustalX, followed by tree generation using a neighbor-joining algorithm using MEGA 6 software (https://www.megasoftware.net). Sequences are labeled by their GenBank accession number. Sequences belonging to lineage III also contain place and year of isolation data. Bootstrap support values are given for each node. Scale bar represents nucleotide substitutions per site. B) Geographic spread of SLEV. A discrete Bayesian phylogeographic reconstruction for SLEV lineage III was made using 11 envelope protein sequences (highlighted cluster in , panel A). We applied a constant-size coalescent tree before the phylogeny and a TNF93 nt substitution model. The Monte Carlo Markov chain model was obtained after 30 million iterations and subsampling every 20.000 iterations. Analyses were made using Beast version 1.8.3 software (http://tree.bio.ed.ac.uk/software/beast/). Numbers over the arrows indicate the probability for the postulated viral dispersion in that pathway. Color of arrows indicates the location origin for the strain introduced.