Volume 25, Number 6—June 2019
Research Letter
Infection with New York Orthohantavirus and Associated Respiratory Failure and Multiple Cerebral Complications
Figure
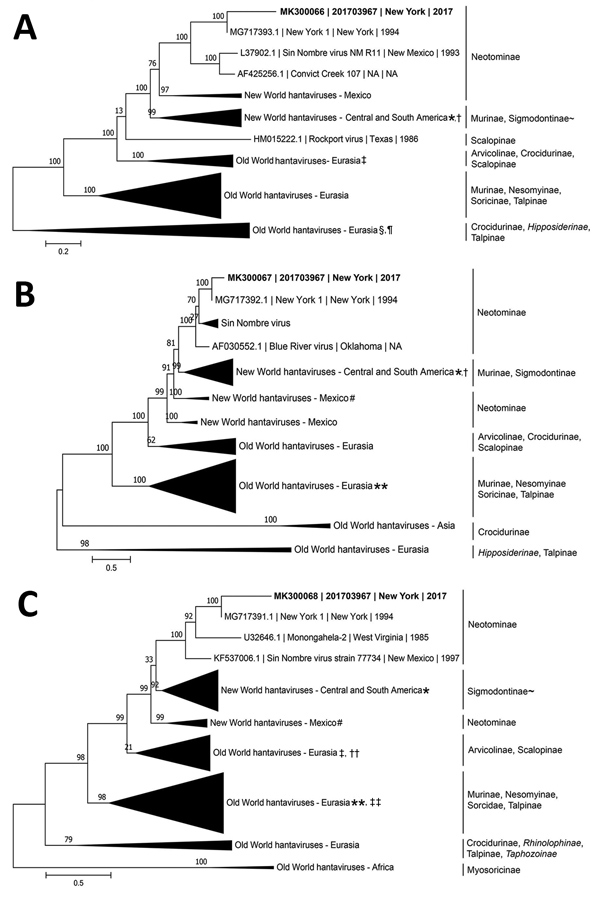
Figure. Inference of phylogenetic relationships for A) large, B) medium, and C) small RNA segments of orthohantaviruses by using representative full-genome sequences in study of infection with New York orthohantavirus and associated respiratory failure and multiple cerebral complications. Partial sequences from the case in New York during 2017 (specimen no. 201703967), are indicated in bold (GenBank accession no. MK300066–8). Genome coverage for the 201703967 small, medium, and large RNA segments are 12%, 30%, and 5%, respectively. Evolutionary history was inferred by using a maximum-likelihood method based on the subtree pruning and regrafting model with the general time-reversible + gamma (n = 4) nucleotide substitution model, and branch support was calculated by using the approximate likelihood ratio test method. Branch lengths are measured in number of nucleotide substitutions per site (scale bars). Clades are collapsed by region for clarity, and clades of interest are annotated with reservoir subfamily. Clades with incomplete reservoir information are annotated with ~, and bat subfamilies are shown in italics. Sequences collected outside a specified geographic region are denoted by a symbol. *Black Creek Canal virus, USA; †Bayou virus, USA; ‡Prospect Hill virus, USA; §Kilimanjaro virus, Tanzania; ¶Uluguru virus, Tanzania; #Limestone Canyon virus, USA; **Sangoussa virus, Guinea; ††Rockport virus, USA; ‡‡Jemez Springs virus, USA.