Volume 26, Number 10—October 2020
Dispatch
Main Routes of Entry and Genomic Diversity of SARS-CoV-2, Uganda
Figure 2
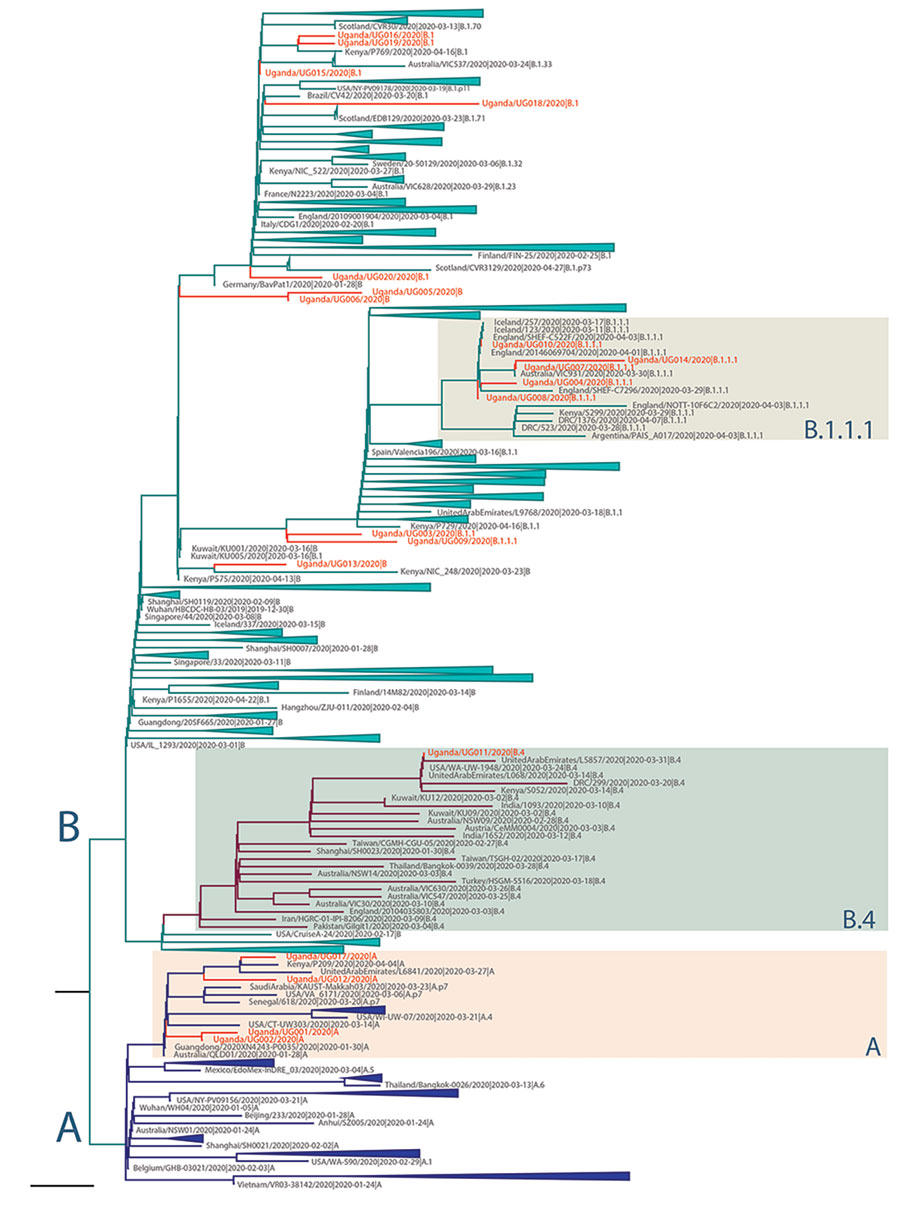
Figure 2. Maximum-likelihood phylogenetic tree of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomes in Uganda. The full SARS-CoV-2 genomes used for phylogenetic lineage nomenclature (A. Rambaut et al., unpub. data, https://doi.org/10.1101/2020.04.17.046086) as defined on May 19, 2020, were retrieved from GISAID (http://www.gisaid.org) (8). Identical sequences were removed, and a total of 395 global representative sequences from each phylogenetic lineage type were selected for further phylogenetic analyses. The reported Uganda sequences, combined with the global SARS-CoV-2 sequences, were aligned by using MAFFT (9) and untranslated regions at 5′ and 3′ were trimmed. Maximum-likelihood phylogenetic tree was constructed in RAxML (10), under the general time-reversible plus gamma distribution model as best-fitted substitution model determined by IQ-TREE (11) and run for 100 pseudo-replicates. The resulting tree was visualized in Figtree (12) and rooted at the point of splitting lineage A and B. Scale bar indicates 6 × 10–5 nucleotide substitutions per site. The branch length is drawn to the scale of nucleotide substitutions per site. The Uganda genomes are indicated in red. The 2 major lineages of SARS-CoV-2 (A and B) are indicated to the left of the tree; the main groups of the Uganda genomes (A, B1.1.1, B4) are indicated by colored boxes to the right of the tree.
References
- Edward C. Holmes, Yong-Zhen Zhang EC. Initial genome release of novel coronavirus [cited 2020 May 7]. http://virological.org/t/initial-genome-release-of-novel-coronavirus/319
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al.; China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–33. DOIPubMedGoogle Scholar
- Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med. 2020;382:1199–207. DOIPubMedGoogle Scholar
- Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–81. DOIPubMedGoogle Scholar
- Johns Hopkins University Center for Systems Science and Engineering. COVID-19 dashboard. 2020 [cited 2020 May 7]. https://www.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6
- World Health Organization. COVID-19 cases top 10,000 in Africa. 2020 [cited 2020 May 7]. https://www.afro.who.int/news/covid-19-cases-top-10-000-africa
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:25. DOIPubMedGoogle Scholar
- Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. DOIPubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. DOIPubMedGoogle Scholar
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Josh B. Singer, Gifford R, Cotten M, Robertson DL. CoV-GLUE project. 2020 [cited 2020 May 7]. http://cov-glue.cvr.gla.ac.uk
1These first authors contributed equally to this article.