Volume 26, Number 2—February 2020
Dispatch
Rapid Nanopore Whole-Genome Sequencing for Anthrax Emergency Preparedness
Cite This Article
Citation for Media
Abstract
Human anthrax cases necessitate rapid response. We completed Bacillus anthracis nanopore whole-genome sequencing in our high-containment laboratory from a human anthrax isolate hours after receipt. The de novo assembled genome showed no evidence of known antimicrobial resistance genes or introduced plasmid(s). Same-day genomic characterization enhances public health emergency response.
Bacillus anthracis causes anthrax, a deadly infectious disease, and is found worldwide, including areas of the United States. Naturally occurring anthrax outbreaks are reported annually in wild and domestic grazing animals, but human transmission is rare (1). Deliberate misuse of B. anthracis as a bioweapon could pose an immediate risk to human populations. In such instances, a timely response is critical to reduce morbidity and mortality rates.
After the anthrax incidents during 2001, the Centers for Disease Control and Prevention (CDC) published medical countermeasure recommendations for human anthrax treatment and postexposure prophylaxis using antimicrobial drugs, including amoxicillin, ciprofloxacin, doxycycline, levofloxacin, and penicillin (2). Most B. anthracis strains are susceptible to antimicrobial drugs; however, naturally occurring and engineered antimicrobial-resistant strains have been reported (3–5). Laboratory antimicrobial susceptibility testing (AST) by broth microdilution (BMD) remains the standard method to determine MIC values but requires >16 hours before results are available. During an anthrax emergency, rapid genomic characterization of the implicated B. anthracis strain(s) could identify sequences associated with drug resistance.
Single-nucleotide mutations in chromosomal B. anthracis quinolone resistance–determining regions of gyrA, gyrB, parC, and parE genes can lead to ciprofloxacin resistance, and gene acquisition can lead to tetracycline and doxycycline resistance (3,4,6). Penicillin resistance can result from a chromosomal mutation in the antisigma factor gene, rsiP (5). Most B. anthracis strains carrying this signature rsiP mutation are resistant to penicillin and amoxicillin (5,7,8). Detection of known antimicrobial resistance (AMR) mutations or other novel gene insertions and deletions (indels) in the clonal B. anthracis genome signals genetic anomalies and could influence treatment and postexposure prophylaxis strategies.
Whole-genome sequencing (WGS) can identify gene indels, mutations, or previously undescribed genetic elements, including extrachromosomal plasmid DNA. However, common short-read sequencing (SRS) technologies have difficulty resolving bacterial genome structure because de novo assemblies yield multiple contigs. Long-read nanopore sequencing with the MinION device (Oxford Nanopore Technologies, https://nanoporetech.com) can resolve repetitive sequences and structural genomic rearrangements and enables complete bacterial genome finishing (9). Although MinION data are error-prone, especially in homopolymeric regions (10), compared with Illumina (https://www.illumina.com)–based SRS, it is available immediately during the sequencing run, enabling rapid assembly and analysis. The technology enables real-time sequencing, including direct pathogen identification from patient specimens, and holds the promise for future point-of-care applications that speed laboratory results reporting (11,12). Portable WGS instruments are advantageous for laboratories with limited space and remove the need to transfer DNA out of high-containment laboratories for sequencing, mitigating exposure risks to personnel (13).
CDC described a rapid nanopore sequencing approach and custom bioinformatics pipeline for B. anthracis that yielded complete chromosome and plasmid assemblies, and detected known AMR genes and mutations in avirulent laboratory strains (13). On the morning of August 2, 2019, our laboratory received a B. anthracis culture isolate (Ba0914) from a naturally occurring human anthrax case in Texas. Same-day laboratory WGS and bioinformatics analysis were performed. This study describes the laboratory work and demonstrates the usefulness of rapid WGS to inform time-sensitive public health responses.
All laboratory work with the B. anthracis isolate and nanopore sequencing was performed inside a class II type A2 biological safety cabinet located in a US Federal Select Agent Program registered Biosafety Level 3 laboratory. We performed rapid nanopore sequencing as described by Gargis et al. (13), including silica membrane–based genomic DNA (gDNA) extraction, except that a bead-beating step was added to speed cell lysis. We extracted Ba0914 gDNA from colonies of an overnight agar culture in 75 min. Within the next hour, fluorometer and microvolume spectrophotometer measurements confirmed that the gDNA extraction was suitable for nanopore sequencing. We prepared a nanopore DNA sequencing library (Rapid Barcoding Sequencing Kit SQK-RBK004; Oxford Nanopore Technologies) and sequencing began <45 min later (MinKNOW, version 18.12.6; Oxford Nanopore Technologies).
Within ≈10 min of sequencing, nanopore data were ready for blastn analysis (https://blast.ncbi.nlm.nih.gov), which identified a 13.5-kb read with >91.5% sequence homology with B. anthracis. Approximately 120,000 live basecalled reads (average length 4,089 nt, average quality score/read 14.8) were generated in <5 hours of nanopore sequencing. We performed de novo genome assembly (Flye version 2.5; https://github.com) by using the first 120,000 nanopore reads and error-corrected with Medaka version 0.6.1 (https://pypi.org).
Figure 1
Figure 1. Time required to detect antimicrobial resistance markers in Bacillus anthracis strain Ba0914 by using WGS and summary of assembly results. A) Comparison of time to complete rapid nanopore (MinION) and short-read...
Figure 2
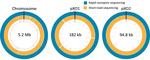
Figure 2. Circular maps of the whole-genome–sequenced Bacillus anthracis Ba0914 chromosome and 2 plasmids, pXO1 and pXO2, assembled by using rapid nanopore sequencing and short-read sequencing. (Maps are not to scale.)
The Ba0914 genome assembly contained single contigs for the chromosome and each plasmid, pXO1 and pXO2 (Figures 1, 2) with >54X average depth of coverage and shared >99.75% identity to the Ames Ancestor strain (GenBank accession no. AE017334) (Table). Thousands of indels and nearly 700 mismatches were detected (Table). Conventional BMD testing also began on August 2, 2019, according to guidelines of the Clinical and Laboratory Standards Institute (14), and susceptibility results were ready the following day.
We also performed Illumina-based SRS. We extracted DNA as described by Gargis et al. (7) and prepared the sequencing library (Nextera DNA Flex Library Kit; Illumina) for paired-end 2 × 150–bp sequencing by using the iSeq 100 (Illumina). We performed read filtering and assembly as described (15). The SRS-based assembly contained 32 more contigs than nanopore but with higher depths of coverage for the chromosome and plasmids (Table). Alignment to the Ames Ancestor reference strain yielded >99.9% genome identity, with fewer indels and mismatches. SRS-based approaches result in lower error rates and can more reliably detect single-nucleotide polymorphisms in B. anthracis, especially in homopolymeric regions, but a same-day laboratory workflow is currently not feasible (Figure 1, panel A). An alignment plot showed gaps in coverage of the chromosome (0.1%) and pXO1 (1.4%) caused by incomplete SRS-based assemblies (Figure 2). Alignment of the nanopore-based assembly to the SRS-based assembly resulted in >99.8% identity but with thousands of indels (Table). All sequencing data were submitted to GenBank under accession no. SAMN12588378.
Only 45 min of bioinformatics analysis (Pima version 01, https://github.com) using 120,000 basecalled nanopore reads was sufficient to assemble and confirm the absence of known AMR genes/markers associated with resistance to quinolones and tetracyclines (Figure 1, panel B) (13). We detected no mutations in gyrA, gyrB, parC, or parE genes, identified no AMR genes contained in the Resfinder database (https://cge.cbs.dtu.dk), and found no unexpected plasmids. SRS and bioinformatics analysis yielded analogous details about the AMR markers in Ba0914 (Figure 1, panel B). Only the SRS-based assembly, and not the nanopore assembly, was reliable for sequencing rsiP. Strain Ba0914 lacked mutations in the homopolymeric rsiP region that can confer penicillin resistance. Sequencing of regions containing repetitive nucleotide bases is a known limitation of nanopore technology and, consequently, detection of the rsiP mutation was excluded from AMR bioinformatics analysis.
Although genetic analysis is useful for detection of known AMR genes/markers in the B. anthracis genome, phenotypic susceptibility testing by BMD remains essential to detect functional resistance (7,13). By using the conventional BMD method, we found that strain Ba0914 was susceptible to ciprofloxacin, levofloxacin, tetracycline, doxycycline, penicillin, and amoxicillin.
Real-time sequencing of the biothreat pathogen B. anthracis in a high-containment laboratory demonstrated the speed and usefulness of a rapid, portable nanopore sequencer during an emergency. Long-read sequencing could detect B. anthracis–specific DNA sequence from the culture isolate after only 3.5 hours. Although the nanopore-based assembly was error-prone when compared with the SRS-based assembly, as few as 8.5 hours would be required to find evidence of known AMR genes/markers or engineering, including gene insertions and extrachromosomal plasmids from B. anthracis. Although conventional AST remains essential for characterizing functional antimicrobial resistance in B. anthracis, nanopore sequencing provided same-day, on-site genomic characterization useful for an anthrax emergency response.
Dr. McLaughlin is a scientist at the Biodefense Research and Development Laboratory, Division of Preparedness and Emerging Infections, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA. Her primary research interests are development and evaluation of phenotypic and whole-genome sequencing–based assays to characterize biological threat agents, with an emphasis on rapid detection of antimicrobial resistance in bacteria.
Acknowledgments
We thank scientists in the Zoonoses and Select Agent Laboratory, Division of High-Consequence Pathogens and Pathology, National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), CDC, for performing short-read sequencing; scientists in the Antimicrobial Resistance and Characterization Laboratory, Division of Healthcare Quality Promotion, NCEZID, CDC, for performing conventional BMD AST; and scientists in the Biodefense Research and Development Laboratory, Division of Preparedness and Emerging Infections, NCEZID, CDC, for performing nanopore sequencing.
This study was supported by the CDC Center for Preparedness and Response.
References
- Carlson CJ, Kracalik IT, Ross N, Alexander KA, Hugh-Jones ME, Fegan M, et al. The global distribution of Bacillus anthracis and associated anthrax risk to humans, livestock and wildlife. Nat Microbiol. 2019;4:1337–43. DOIPubMedGoogle Scholar
- Hendricks KA, Wright ME, Shadomy SV, Bradley JS, Morrow MG, Pavia AT, et al. Workgroup on Anthrax Clinical Guidelines. Centers for disease control and prevention expert panel meetings on prevention and treatment of anthrax in adults. Emerg Infect Dis. 2014;20:
e130687 . DOIGoogle Scholar - Price LB, Vogler A, Pearson T, Busch JD, Schupp JM, Keim P. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob Agents Chemother. 2003;47:2362–5. DOIPubMedGoogle Scholar
- Pomerantsev AP, Shishkova NA, Marinin LI. [Comparison of therapeutic effects of antibiotics of the tetracycline group in the treatment of anthrax caused by a strain inheriting tet-gene of plasmid pBC16] [in Russian]. Antibiot Khimioter. 1992;37:31–4.PubMedGoogle Scholar
- Ross CL, Thomason KS, Koehler TM. An extracytoplasmic function sigma factor controls beta-lactamase gene expression in Bacillus anthracis and other Bacillus cereus group species. J Bacteriol. 2009;191:6683–93. DOIPubMedGoogle Scholar
- Weigel LM, Sue D, Michel PA, Kitchel B, Pillai SP. A rapid antimicrobial susceptibility test for Bacillus anthracis. Antimicrob Agents Chemother. 2010;54:2793–800. DOIPubMedGoogle Scholar
- Gargis AS, McLaughlin HP, Conley AB, Lascols C, Michel PA, Gee JE, et al. Analysis of whole-genome sequences for the prediction of penicillin resistance and β-lactamase activity in Bacillus anthracis. mSystems. 2018;3:e00154–18. DOIPubMedGoogle Scholar
- Ågren J, Finn M, Bengtsson B, Segerman B. Microevolution during an Anthrax outbreak leading to clonal heterogeneity and penicillin resistance. PLoS One. 2014;9:
e89112 . DOIPubMedGoogle Scholar - Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb Genom. 2017;3:
e000132 . DOIPubMedGoogle Scholar - Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–51. DOIPubMedGoogle Scholar
- Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530:228–32. DOIPubMedGoogle Scholar
- Greninger AL, Naccache SN, Federman S, Yu G, Mbala P, Bres V, et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015;7:99. DOIPubMedGoogle Scholar
- Gargis AS, Cherney B, Conley AB, McLaughlin HP, Sue D. Rapid detection of genetic engineering, structural variation, and antimicrobial resistance markers in bacterial biothreat pathogens by nanopore sequencing. Sci Rep. 2019;9:13501. DOIPubMedGoogle Scholar
- Clinical and Laboratory Standards Institute. Methods for antimicrobial dilution and disc susceptibility testing of infrequently isolated or fastidious bacteria, 3rd ed. M45. Wayne (PA): The Institute; 2016.
- Nouioui I, Klenk H-P, Igual JM, Gulvik CA, Lasker BA, McQuiston JR. Streptacidiphilus bronchialis sp. nov., a ciprofloxacin-resistant bacterium from a human clinical specimen; reclassification of Streptomyces griseoplanus as Streptacidiphilus griseoplanus comb. nov. and emended description of the genus Streptacidiphilus. Int J Syst Evol Microbiol. 2019;69:1047–56. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: January 08, 2020
Table of Contents – Volume 26, Number 2—February 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Heather P. McLaughlin, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop H17-5, Atlanta, GA 30329-4027, USA
Top