Volume 26, Number 6—June 2020
Synopsis
Genomic Epidemiology of 2015–2016 Zika Virus Outbreak in Cape Verde
Cite This Article
Citation for Media
Abstract
During 2015–2016, Cape Verde, an island nation off the coast of West Africa, experienced a Zika virus (ZIKV) outbreak involving 7,580 suspected Zika cases and 18 microcephaly cases. Analysis of the complete genomes of 3 ZIKV isolates from the outbreak indicated the strain was of the Asian (not African) lineage. The Cape Verde ZIKV sequences formed a distinct monophylogenetic group and possessed 1–2 (T659A, I756V) unique amino acid changes in the envelope protein. Phylogeographic and serologic evidence support earlier introduction of this lineage into Cape Verde, possibly from northeast Brazil, between June 2014 and August 2015, suggesting cryptic circulation of the virus before the initial wave of cases were detected in October 2015. These findings underscore the utility of genomic-scale epidemiology for outbreak investigations.
Zika virus (ZIKV), first discovered in Uganda in 1947 and sporadically found in Africa and Asia, was long believed to only cause mild disease in humans (1). ZIKV isolates are classified into 1 of 2 lineages, representing the African and Asian genotypes. ZIKVs of the African lineage have been isolated from many regions of Africa (2), mostly through entomologic investigations, and serologic evidence suggests that ZIKV infections in humans are frequent (3). However, until the 2000s, the virus had seldom been detected in humans. The Asian lineage has spread throughout the Pacific, causing outbreaks in humans in Yap, Federated States of Micronesia, in 2007 and in French Polynesia during 2013–2014, where an association with neurologic afflictions was first detected (4). Zika cases were first reported in Brazil in May 2015, and from there, the virus quickly spread to most of the Americas (5). The high number of cases led to the discovery of an association between congenital ZIKV infection and neonatal neurologic complications, particularly microcephaly (6,7).
Figure 1
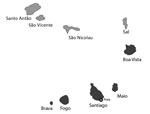
Figure 1. Locations of suspected Zika cases (dark gray shading), Cape Verde, 2015–2016. Only 2 cases on Boa Vista were confirmed, and those might have been imported.
In October 2015, an epidemic of rash, conjunctivitis, and arthralgia was noted by physicians in Praia, the capital of Cape Verde, an archipelago nation located in the Atlantic Ocean, west of the coast of Senegal. Blood samples sent to the regional reference laboratory of the Institut Pasteur de Dakar (Dakar, Senegal) confirmed the epidemic involved ZIKV infection. By the end of the outbreak in May 2016, a total of 7,580 suspected Zika cases and 18 microcephaly cases were reported in the 4 most densely populated southern islands of the Cape Verde archipelago (Brava, Fogo, Maio, and Santiago; Figure 1) (8). Overall, ≈50% of confirmed microcephaly cases were linked to reports of Zika-related symptoms in the mother during the first trimester of gestation (8).
Sample Collection
In October 2015, Cape Verde reported a ZIKV outbreak and initiated a surveillance system to investigate the circulation of the virus in the country. All healthcare facilities were alerted to report suspected Zika cases according to the case definition of rash with or without fever and >1 of the following symptoms: conjunctivitis, headache, pruritus, arthralgia, myalgia, diarrhea, vomiting, adenopathy, or retro-orbital pain. In total, 1,226 sample sets (including blood and sometimes matching urine) of the original 7,580 sample sets from patients with suspected ZIKV infections (9) were sent to the Virology Laboratory at the Achadinha Health Center (Praia, Cape Verde) for ZIKV diagnosis. Only a fraction of the original sample set was sent because ZIKV testing was performed at the discretion of each healthcare facility’s medical practitioners. Staff also sent samples from patients not fitting the Zika case definition (i.e., patients with only rash or only fever) for Zika testing.
Ethics
In this study, we used samples collected as part of approved ongoing surveillance conducted by the Institut Pasteur de Dakar (a World Health Organization Collaborating Centre for Arboviruses and Haemorrhagic Fever Reference and Research). All samples from humans were de-identified before we performed virus characterization and analyses; thus, no patient information can be reported.
Molecular Tests
We tested acute serum samples (obtained <5 days after symptom onset; n = 387) and, when available, matched urine samples (n = 82) by quantitative reverse transcription PCR (qRT-PCR). We extracted RNA from serum or urine samples using the QIAamp Viral RNA Mini Kit (QIAGEN, https://www.qiagen.com) according to the manufacturer’s recommendations and performed a 1-step real-time PCR assay (10) on an ABI7500 instrument (Applied Biosystems, https://www.thermofisher.com) using the QuantiTect Probe RT-PCR Kit (QIAGEN).
Serologic Tests
We tested serum samples (collected <10 days after symptom onset; n = 1,226) by ELISA for ZIKV IgM and IgG. For ZIKV antibody–positive samples, we tested for antibodies against other flaviviruses (yellow fever virus [YFV], dengue virus [DENV], and West Nile virus [WNV]) endemic in the West Africa region to rule out cross-reactions.
For the IgM ELISA, we coated 96-well microtiter plates with a monoclonal IgM capture antibody (goat anti-human IgM; KPL, https://www.seracare.com) in carbonate bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. After washing the plate 3 times with phosphate-buffered saline plus 0.05% tween, we added heat-inactivated (56°C, 30 min) patient serum samples and controls (all diluted 1:100 in phosphate-buffered saline plus 0.05% tween and 1% milk powder) in duplicate into plate wells and incubated at 37°C for 1 h. We washed wells 3 times; added ZIKV, DENV, WNV, or YFV antigens into plate wells; and incubated the plate for 1 h. After 3 washes, we added ZIKV-, DENV-, WNV-, or YFV-specific immune ascites from mice to each well and incubated for 1 h at 37°C. After 3 washes, we added peroxidase-labeled antibody specific to mouse IgG for 1 h at 37°C. Last, we added a tetramethylbenzidine substrate to the IgM conjugate complex and stopped the color reaction using a sulfuric acid solution. For the indirect IgG ELISA, we captured ZIKV antigen on 96-well plates coated with ZIKV-specific mouse hyperimmune ascitic fluid. We added patient serum samples (1:100) and then horseradish peroxidase–conjugated goat anti-human IgG. We considered serum samples positive if the optical density at 450 nm was >0.20 above the negative serum sample average and the ratio between the sample and the negative control was >2.
We analyzed samples positive for ZIKV IgM or IgG by ELISA for ZIKV-neutralizing antibodies using the plaque reduction neutralization test (PRNT) as described by De Madrid and Porterfield (11). In brief, we mixed 2-fold serial dilutions of serum samples (starting at 1:10) with equal volumes of medium containing 800 PFU/mL of the ZIKV reference strain MR766 and incubated for 1 h at 37°C. We then used serum–virus mixtures to infect Vero cell monolayers in 24-well plates. After 1 h at 37°C, we covered cells with DMEM containing 2% FBS and 0.4% carboxymethylcellulose and incubated for 4 days. We determined neutralizing antibody titers using an 80% cutoff value and classified samples as positive if their titers were ≥20 IU/mL.
Untargeted Sequencing
We treated RNA samples first with Turbo DNase (Ambion, https://www.thermofisher.com) and then depleted host rRNA using the NEBNext rRNA Depletion Kit (New England Biolabs, https://www.neb.com). We used rRNA-depleted RNA samples for cDNA synthesis using random primers and Superscript IV (ThermoFisher, https://www.thermofisher.com) and used cDNA for library preparation (Nextera XT DNA Library Prep Kit; Illumina, https://www.illumina.com). We sequenced libraries on an Illumina NextSeq500.
Amplicon-Based Sequencing
We sequenced amplicons from 3 samples following the protocol described by Quick et al. (12). We prepared sequencing libraries using the NEBNext Ultra II DNA Library Prep Kit for Illumina and barcoded with NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) (New England Biolabs for both). We sequenced prepared libraries on an Illumina MiSeq using MiSeq Reagent Kit v3 (600 cycles) at the Biomics platform of Institut Pasteur (Paris, France). We used Trimmomatic (http://www.usadellab.org) to remove adaptor and primer sequences (first 24 nt from 5′ end of reads, which is the maximum length of primers used for multiplexed PCRs). Then, we aligned reads to the complete genome sequence of a 2016 ZIKV isolate from the Dominican Republic (GenBank accession no. KU853012) using the CLC Genomics Suite (QIAGEN). We used SAMtools (http://samtools.sourceforge.net) to sort the aligned bam files and generate alignment statistics. We visually inspected ZIKV-aligned reads using Geneious version 9.1 (https://www.geneious.com) before generating consensus sequences and required a minimum of a 5× read-depth coverage to make a base call. We deposited all sequences we obtained in GenBank (accession nos. MK241415–7).
Sequence Dataset Compilation
To determine the origin of ZIKV in Cape Verde, we created a comprehensive dataset of all publicly available ZIKV sequences from GenBank. We then removed from this dataset all laboratory, patented, African genotype, and pre-2000 Asian genotype isolates. We concatenated all genome fragments of the same isolate to retain 1 representative sequence per isolate. For sequences with incomplete information on sampling time and location in GenBank records, we attempted to retrieve this information from published studies and other online resources. For cases in which the origin of the imported case of ZIKV infection was known, we set the sampling location as the location of origin. Last, we removed sequences that were not linked to a publication or associated with a known sampling time or location. This data pruning resulted in a final dataset of 459 ZIKV sequences. We created a multiple sequence alignment of these data using MAFFT (13) and manually edited the alignment by using AliView (14).
Phylogenetic Inference
We performed Bayesian phylogenetic and phylogeographic analyses using BEAST version 1.10.4 (15). We employed an uncorrelated relaxed-clock model (with rates drawn from an underlying log-normal distribution) (16) and a skygrid coalescent tree prior distribution (17) in combination with a codon-partitioned substitution model (SRD06) (18). We inferred migration history using a discrete phylogeographic model that incorporates a model-averaging procedure (i.e., the Bayesian stochastic search variable selection procedure) (19) to identify the subset of migration flows that adequately explain the diffusion process. We assessed convergence and mixing properties of the Markov chains with Tracer version 1.7 (20) and combined samples of several independent chains post burn-in. We generated maximum clade credibility trees summarizing the combined post burn in Markov chain Monte Carlo samples with the use of TreeAnnotator version 1.10.4 (http://beast.community/programs).
Figure 2

Figure 2. Suspected Zika cases, cases tested for Zika virus (ZIKV) infection, ZIKV antibody–positive cases, and ZIKV RNA–positive cases, Cape Verde, 2015–2016, by epidemiologic week. A) Cases of suspected ZIKV infection (n =...
We report the findings of samples from 1,226 patients with suspected ZIKV infections (Table), as well as patients not fitting the Zika case definition. Samples were tested over the course of the Cape Verde outbreak. The practice of sending samples from cases not fulfilling the strict Zika case definition for ZIKV testing resulted in a surge of tests being performed during the latter part of the outbreak (Figure 2). The peak of acute infections (as detected by IgM ELISA or qRT-PCR) occurred 3 weeks before the peak of suspected cases. The presence of IgG-positive, IgM-negative, qRT-PCR–negative cases in April 2016 indicates that many infections were not detected during their acute phase and highlights the challenge of capturing the timing and extent of outbreaks when a large proportion of the infected population is asymptomatic. Indeed, 80% of ZIKV infections are asymptomatic (21). The early detection of samples positive for ZIKV IgG (confirmed by PRNT) and negative for ZIKV IgM and RNA (including samples with sampling dates before October 2015 that were also received for ZIKV testing though not fitting the Zika case definition) is compatible with the silent circulation of ZIKV before the major wave of reported cases. Although the high population densities of the southern islands (with >50% of the Cape Verde population living on Santiago Island) may explain why these islands experienced intense outbreaks, determining other factors that might have helped prevent further spread of the virus within the archipelago during the outbreak will be of value for future application.
Before this outbreak on the Cape Verde archipelago, no Zika outbreaks or ZIKV-associated microcephaly cases had been reported in Africa, and only the Asian lineage of ZIKV had been found to be associated with neurologic and congenital afflictions. Because Cape Verde is situated in an area (near Senegal) where the African lineage of ZIKV has been regularly detected (22) and the Cape Verde outbreak involved cases of microcephaly, we needed to determine the origin and genotype of the ZIKV that caused this outbreak. We assessed virus origin and genotype through sequencing and phylogenetic analysis of ZIKVs sampled from patients in Cape Verde. Although our attempts at untargeted sequencing with rRNA-depleted samples were unsuccessful, we were able to Sanger sequence a small fragment of the envelope gene, which revealed that the virus was a part of the Asian lineage. We shared this information with the World Health Organization in May 2016.
Figure 3

Figure 3. Maximum clade credibility phylogenetic tree demonstrating migration history of Zika virus (ZIKV) Asian lineage, 2014–2018. A) Phylogeny of 459 ZIKV isolates. The tree base was removed for ease of presentation. Tips...
We subsequently used a published overlapping PCR amplicon scheme to amplify and sequence the ZIKV genomes of preserved samples (12). This procedure enabled us to construct libraries that we could sequence and resulted in us obtaining 3 complete ZIKV genomes. These 3 sequences were obtained from samples acquired in November 2015 (Fogo), December 2015 (Santiago), and April 2016 (Fogo). Bayesian phylogenetic analysis of publicly available ZIKV sequences indicated that all 3 Cape Verde ZIKV genomes were related to the Asian genotype and formed a single, separate, well-supported monophyletic group (posterior probability 1, Figure 3; Appendix Figure 1), confirming our initial inference that was based on a partial envelope gene fragment. Of note, all sequences of Cape Verde ZIKVs contained a unique nonsynonymous substitution in the envelope protein (I756V), and a second amino acid change (T659A) also in the envelope gene was present in 2 of the isolates (those acquired December 2015 and April 2016). Additional work is needed to determine whether these mutations arose by chance or if they facilitate adaptation to local conditions, such as growth in mosquito species of Africa.
Phylogeographic analyses indicated that the Cape Verde ZIKV clade shares a common ancestor with lineages circulating in Brazil (posterior probability 0.798) and is most likely from northeast Brazil (posterior probability 0.574). Both Brazil and Cape Verde are Portuguese-speaking countries, and direct flights between northeast Brazil (Fortaleza) and Cape Verde (Praia) occurred daily during the outbreak. Although our data suggest that the ZIKV responsible for the Cape Verde outbreak originated in northeast Brazil, a lack of genomic sampling from many ZIKV-affected regions in the Americas makes excluding transmission through these other locations impossible. The median estimated date for the most recent common ancestor of the Cape Verde ZIKV clade was June 2015 (95% highest posterior density March 2015–August 2015; Figure 3). The median estimated date of divergence of this clade from the sequences sampled outside Cape Verde was November 2014 (95% highest posterior density July 2014–March 2015). Hence, these results indicate that ZIKV was most likely introduced into Cape Verde between June 2014 and August 2015, clearly before the first wave of documented clinical cases. This estimated virus introduction date is also supported by the finding of persons with ZIKV IgG and neutralizing antibodies early during the outbreak (Figure 2).
This study has 2 main limitations. First, because only a fraction of suspected Zika case samples were tested, bias in the reported distribution of confirmed cases is possible. Second, only 3 ZIKV genome sequences were recovered from this outbreak, perhaps limiting the power of our phylodynamic and phylogeographic analyses.
In conclusion, our analysis indicates that the Asian lineage of ZIKV reached Cape Verde in 2015 and triggered a relatively large epidemic. Of note, the Cape Verde ZIKV clade is distinct from the ZIKV of the 2017 Angola outbreak (Appendix Figure 2), which also involved microcephaly cases (23,24). The introduction of ZIKV into Cape Verde from Brazil and silent circulation in Cape Verde was likely to have occurred before the wave of cases in October 2015, as revealed by both phylogenetic and serologic data. The time scale we report is consistent with those estimated for the initial movement of ZIKV from Brazil to neighboring countries in South America and islands of the Caribbean (Appendix Figure 1) (10,25,26).
Dr. Oumar Faye is a virologist often working on outbreak responses at Institut Pasteur de Dakar in Dakar, Senegal. His research interests focus on the development of diagnostic tools and the characterization of arboviruses and hemorrhagic fever viruses.
Acknowledgments
We thank 2 anonymous reviewers whose suggestions helped to improve this manuscript.
This work was supported by Institut Pasteur de Dakar. P.L. received funding by the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement no. 725422-ReservoirDOCS) and by the Wellcome Trust through project 206298/Z/17/Z. S.L. and B.V. are supported by the Fonds Wetenschappelijk Onderzoek (Belgium). E.S.-L. received funding from the INCEPTION program (Investissements d’Avenir grant ANR-16-CONV-0005) and Institut Pasteur. F.G.-R. is part of the Pasteur-Paris University International doctoral program, BioSPC doctoral school. E.C.H. is funded by an Australian Research Council Australian Laureate Fellowship (FL170100022).
References
- Baud D, Gubler DJ, Schaub B, Lanteri MC, Musso D. An update on Zika virus infection. Lancet. 2017;390:2099–109. DOIPubMedGoogle Scholar
- Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, Diallo M, et al. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis. 2014;8:
e2636 . DOIPubMedGoogle Scholar - Posen HJ, Keystone JS, Gubbay JB, Morris SK. Epidemiology of Zika virus, 1947-2007. BMJ Glob Health. 2016;1:
e000087 . DOIPubMedGoogle Scholar - Cao-Lormeau VM, Blake A, Mons S, Lastère S, Roche C, Vanhomwegen J, et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet. 2016;387:1531–9. DOIPubMedGoogle Scholar
- Pan American Health Organization; World Health Organization. Zika cases and congenital syndrome associated with Zika virus reported by countries and territories in the Americas, 2015–2018, cumulative cases. 2018 Jan 4 [cited 2019 Oct 15]. https://www.paho.org/hq/index.php?option=com_docman&task=doc_download&gid=43296
- de Araújo TVB, Ximenes RAA, Miranda-Filho DB, Souza WV, Montarroyos UR, de Melo APL, et al.; investigators from the Microcephaly Epidemic Research Group; Brazilian Ministry of Health; Pan American Health Organization; Instituto de Medicina Integral Professor Fernando Figueira; State Health Department of Pernambuco. Association between microcephaly, Zika virus infection, and other risk factors in Brazil: final report of a case-control study. Lancet Infect Dis. 2018;18:328–36. DOIPubMedGoogle Scholar
- Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374:951–8. DOIPubMedGoogle Scholar
- Lourenço J, de Lourdes Monteiro M, Valdez T, Monteiro Rodrigues J, Pybus O, Rodrigues Faria N. Epidemiology of the Zika Virus Outbreak in the Cabo Verde Islands, West Africa. PLoS Curr. 2018;10:10. DOIPubMedGoogle Scholar
- Serviço de Vigilância Integrada e Resposta a Epidemias, Ministério da Saúde e da Segurança Social. Boletim informativo seminal, infeção por vírus Zika (ZIKV), ano 2016, (semanas 1 a 21). 2016 [cited 2019 Jul 1]. http://www.minsaude.gov.cv/index.php/documentosite/zika-1/341-boletim-informativo-semanal-da-infecao-por-virus-zika-semana-21-ano-2016/file
- Faria NR, Azevedo RDSDS, Kraemer MUG, Souza R, Cunha MS, Hill SC, et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science. 2016;352:345–9. DOIPubMedGoogle Scholar
- De Madrid AT, Porterfield JS. A simple micro-culture method for the study of group B arboviruses. Bull World Health Organ. 1969;40:113–21.PubMedGoogle Scholar
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017;12:1261–76. DOIPubMedGoogle Scholar
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 2014;30:3276–8. DOIPubMedGoogle Scholar
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:
vey016 . DOIPubMedGoogle Scholar - Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:
e88 . DOIPubMedGoogle Scholar - Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol Biol Evol. 2013;30:713–24. DOIPubMedGoogle Scholar
- Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol. 2006;23:7–9. DOIPubMedGoogle Scholar
- Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLOS Comput Biol. 2009;5:
e1000520 . DOIPubMedGoogle Scholar - Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67:901–4. DOIPubMedGoogle Scholar
- Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med. 2009;360:2536–43. DOIPubMedGoogle Scholar
- Sow A, Loucoubar C, Diallo D, Faye O, Ndiaye Y, Senghor CS, et al. Concurrent malaria and arbovirus infections in Kedougou, southeastern Senegal. Malar J. 2016;15:47. DOIPubMedGoogle Scholar
- Hill SC, Vasconcelos J, Neto Z, Jandondo D, Zé-Zé L, Aguiar RS, et al. Emergence of the Asian lineage of Zika virus in Angola: an outbreak investigation. Lancet Infect Dis. 2019;19:1138–47. DOIPubMedGoogle Scholar
- Sassetti M, Zé-Zé L, Franco J, Cunha JD, Gomes A, Tomé A, et al. First case of confirmed congenital Zika syndrome in continental Africa. Trans R Soc Trop Med Hyg. 2018;112:458–62. DOIPubMedGoogle Scholar
- Faria NR, Quick J, Claro IM, Thézé J, de Jesus JG, Giovanetti M, et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. 2017;546:406–10. DOIPubMedGoogle Scholar
- Thézé J, Li T, du Plessis L, Bouquet J, Kraemer MUG, Somasekar S, et al. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe. 2018;23:855–864.e7. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: April 29, 2020
1These first authors contributed equally to this article.
2These senior authors contributed equally to this article.
Table of Contents – Volume 26, Number 6—June 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Etienne Simon-Loriere, Institut Pasteur, 28 rue du Docteur Roux, 75724 Paris CEDEX 15, France
Top