Volume 27, Number 1—January 2021
Research
Viral Metagenomic Analysis of Cerebrospinal Fluid from Patients with Acute Central Nervous System Infections of Unknown Origin, Vietnam
Cite This Article
Citation for Media
Abstract
Central nervous system (CNS) infection is a serious neurologic condition, although the etiology remains unknown in >50% of patients. We used metagenomic next-generation sequencing to detect viruses in 204 cerebrospinal fluid (CSF) samples from patients with acute CNS infection who were enrolled from Vietnam hospitals during 2012–2016. We detected 8 viral species in 107/204 (52.4%) of CSF samples. After virus-specific PCR confirmation, the detection rate was lowered to 30/204 (14.7%). Enteroviruses were the most common viruses detected (n = 23), followed by hepatitis B virus (3), HIV (2), molluscum contagiosum virus (1), and gemycircularvirus (1). Analysis of enterovirus sequences revealed the predominance of echovirus 30 (9). Phylogenetically, the echovirus 30 strains belonged to genogroup V and VIIb. Our results expanded knowledge about the clinical burden of enterovirus in Vietnam and underscore the challenges of identifying a plausible viral pathogen in CSF of patients with CNS infections.
Worldwide, the annual incidence of acute encephalitis in nonoutbreak settings during 1983–2000 ranged from 0.07 to 12.6 cases/100,000 population (1). According to the World Health Organization, meningitis caused 379,000 deaths and encephalitis caused 150,000 deaths globally in 2015 (2). As a consequence, central nervous system (CNS) infection is a leading cause of years lived with disability in low-income countries (3).
More than 100 known pathogens can cause CNS infections (1). However, the distribution of CNS infection pathogens is geographically dependent and has been shaped by the emergence of novel viruses. In Asia, Nipah virus and enterovirus A71 have been recognized as emerging neurotropic pathogens over the past few decades. In 1999, West Nile virus arrived in the United States and since then has established endemic circulation (4).
Despite recent advances in molecular diagnostics, especially sensitive virus-specific PCR, encephalitis cases of unknown origin remain a substantial problem. Worldwide, ≈50% of patients with CNS infections have no etiology identified (1,5,6).
Over the past decade, metagenomic next-generation sequencing (mNGS) has emerged as a sensitive hypothesis-free approach for detection of pathogens (especially viruses) in clinical samples (7). However, in resource-limited settings like Southeast Asia and Vietnam, a limited number of mNGS studies examining known and unknown viruses in cerebrospinal fluid (CSF) samples from patients with CNS infections have been conducted, even though in this tropical region of the world, novel viruses are likely to emerge (P. Zhou et al., unpub. data, https://doi.org/10.1101/2020.01.22.914952), and diverse CNS infection pathogens have been documented. Collectively, improving our knowledge about viral causes of CNS infections is essential for clinical management and development of intervention strategies. In this study, by using a mNGS approach, we set out to search for known and unknown viruses in CSF samples collected from patients in Vietnam with CNS infections of unknown causes who were enrolled in a hospital-based surveillance study conducted during 2012–2016.
Clinical Study and Selection of CSF Samples for mNGS Analysis
The study used CSF samples collected from patients with suspected CNS infection enrolled in a hospital-based surveillance program conducted in Vietnam during December 2012–October 2016 (5). The study was conducted as part of the Vietnam Initiative on Zoonotic Infections (VIZIONS) project (5), and patient recruitment was carried out at 7 provincial hospitals across Vietnam. After collection, as per the study protocol, all CSF samples were tested for a range of pathogens by using the diagnostic work-up of the clinical study (Appendix Table 1). The remaining volume of the CSF samples were stored at −80°C for further testing.
Figure 1
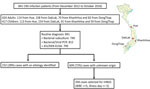
Figure 1. Flowchart overview of diagnostic results for study of patients with suspected central nervous system infections admitted to 4 of 7 provincial hospitals, Vietnam, December 2012–October 2016. Inset map indicates places...
We focused our metagenomic analysis on patients of unknown origin from 4 provincial hospitals in central (Hue and KhanhHoa), highland (DakLak), and southern (DongThap) Vietnam (Figure 1), representing 3 distinct geographic areas in Vietnam. To increase the chance of detecting a virus in the CSF samples, we only selected patients with CSF leukocyte counts >5 cells/mm3 and an illness duration <5 days.
mNGS Assay
mNGS assay was carried out as previously described (8). Before viral nucleic acid (NA) isolation, 100 μL of each CSF sample was treated with Turbo DNase (Ambion, Life Technology, ThermoFisher, https://www.thermofisher.com) and RNase I enzyme (Ambion). Then viral NA was isolated using a QIAamp viral RNA kit (QIAGEN GmbH, https://www.qiagen.com), and recovered in 50 μL of elution buffer provided with the extraction kit. Double-stranded DNA was synthesized from the isolated viral NA by using a set of 96 nonribosomal primers (FR26RV–Endoh primers) and then was randomly amplified by using the FR20RV primer (5′-GCCGGAGCTCTGCAGATATC-3′). Finally, the amplified product was subjected to a library preparation step by using Nextera XT sample preparation kit (Illumina, https://www.illumina.com), following the manufacturer’s instructions, and sequenced by using a MiSeq reagent kit, version 3 (600 cycles) (Illumina) in a MiSeq platform (Illumina).
mNGS Data Analysis
Potential viral reads were identified by using an in-house viral metagenomic pipeline running on a 36-node Linux cluster as described previously (9). In brief, after duplicate reads and reads belonging to human or bacterial genomes were filtered out, the remaining reads were assembled de novo. The resulting contigs and singlet reads were then aligned against a customized viral proteome database by using an approach based on BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Next, the candidate viral reads were aligned against a nonredundant nonvirus protein database to remove any false-positive reads (i.e., reads with expected values higher than those against viral protein databases). Any virus-like sequence with an expected value <0.00001 was considered a significant hit. Finally, a reference-based mapping approach (Genious 8.1.5; Biomatters, https://www.geneious.com) was used to assess the levels of identity and genome coverage of the corresponding viruses.
PCR Confirmatory Testing of mNGS Results
PCR assays were conducted to confirm mNGS hits for each specific virus identified from the viral metagenomic pipeline. Depending on availability of CSF, the PCR confirmations were performed either on leftover NA or newly extracted NA. A viral mNGS result was considered positive only if it was subsequently confirmed by PCR analysis of the original NA samples. The nucleotide sequences of primers and probes used for PCR confirmatory testing are shown in Appendix Table 2 (8).
Serotype Identification and Phylogenetic Analysis
For enterovirus serotype determination based on the obtained sequences generated by viral mNGS, we used a publicly available genotyping tool (10). To determine the relationship between enterovirus strains we sequenced and global strains, we first performed pairwise alignment by using the ClustalW tool in Geneious 8.1.5, and then reconstructed a maximum-likelihood phylogenetic tree by using IQ Tree 1.4.3 (11). A similar phylogenetic approach was used for other viruses. The generated sequences of this study were submitted to GenBank (accession no. PRJNA561465).
Ethics
The study was approved by the corresponding institutional review broad of local hospitals in Vietnam, where the patients were enrolled, and the Oxford Tropical Ethics Committee. Informed consent was obtained from each study participant or a legal guardian.
CSF Samples Available for mNGS Analysis
From the clinical study described previously, a total of 841 patients were enrolled from Hue, Khanh Hoa, Dak Lak, or Dong Thap provincial hospitals. Of these, 609/841 (72%) patients had no etiology identified. The etiologic profiles of the patients in whom a pathogen was detected will be reported separately. Of the patients in whom a pathogen was not identified, 204 met our selection criteria, and their CSF samples were included for viral mNGS analysis (Figure 1).
Baseline Characteristics of the Included Patients
The baseline characteristics and outcome of the 204 study patients are described in Table. Male patients were predominant. A substantial proportion of the patients were seriously ill; fatal outcome was recorded in 22 (11%), whereas incomplete recovery was recorded in 17% (n = 35) and deterioration (reflected by being transferred to other hospitals) in 16.5% (n = 34).
General Description of mNGS Results
Figure 2

Figure 2. Number of cerebrospinal fluid samples with detected viruses by metagenomic next-generation sequencing and then confirmed by virus-specific PCR or reverse-transcription PCR, Vietnam, December 2012–October 2016. Samples were collected from patients...
A total of 204 CSF samples were subjected to 3 NGS runs, and 108 million reads were obtained (median number of reads per sample 445,412 [range 430–908,890]). Of these, viral reads accounted for 0.64% (n = 692,731; median number of reads per sample 2,001 [range 4–268,933]). Excluding common contaminants and commensal viruses such as torque teno virus, which are not reported in this article, sequences related to a total of 8 distinct viral species were identified in 107/204 (52.4%) patients. These viruses are either known to be infectious to humans (e.g., enteroviruses, rotavirus, molluscum contagiosum virus [MCV], human papillomavirus, HIV, and hepatitis B virus [HBV]) or are without evidence of human infections besides previous detection in sterile human samples (e.g., cyclovirus-VN and gemycircularvirus) (Figure 2).
mNGS Result Assessment by Specific PCR Analysis
After virus-specific PCR confirmatory testing, the proportion of patients in whom a virus was found by mNGS was reduced from 53% (108/204) to 14.7% (30/204). Accordingly, the number of virus species was reduced from 8 to 5 (Figure 2); enteroviruses were the most common virus detected, accounting for 11.3% (23/204) of the included patients, followed by HBV (n = 3), HIV (n = 2), gemycircularvirus, and MCV (1 each) (Figure 2). Because of the focus of our study and the unavailability of the PCR assays, confirmatory testing for human papillomavirus was not performed.
Characteristics of the 23 Enterovirus-Infected Patients
All 23 enterovirus-infected patients were admitted to hospitals from the central or highland areas (Table), and none were from Dong Thap Province. Male patients were slightly predominant, accounting for 56%. Notably, the enterovirus-infected patients were younger than those who were mNGS-negative (Table). At discharge, incomplete recovery or transfer to other hospitals because of disease deterioration were recorded in 21.7% (Table).
Figure 3

Figure 3. Temporal distribution of enterovirus cases detected from cerebrospinal fluid samples of patients with suspected central nervous system infection by metagenomic next-generation sequencing and RT-PCR, Vietnam, December 2012–October 2016. Enterovirus RT-PCR...
Enterovirus cases were not detected during January 2015–December 2016. During 2013 and 2014, two main peaks were observed during March–July and September–December (Figure 3, panel A); cases from Dak Lak and Khanh Hoa contributed to the first peak (Figure 3, panels B and C), and cases from Khanh Hoa and Hue contributed to the second (Figure 3, panels C and D). The general baseline characteristics of patients with HBV, gemycircularvirus, and MCV are shown in Appendix Table 3.
Genetic Characterization of Enteroviruses and Gemycircularvirus
Figure 4
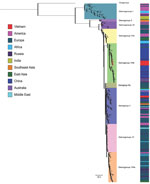
Figure 4. Phylogenetic tree of 298 complete viral protein 1 sequences of echovirus 30 (876 nt) isolated from cerebrospinal fluid samples of patients with suspected central nervous system infection, Vietnam, December 2012–October...
mNGS generated sufficient sequence information for an enterovirus serotyping assessment in 11/23 cases. Subsequently, results of serotyping analysis based on the NGS sequences showed that echovirus 30 (E30) was the most common serotype detected (n = 9, 39% of enteroviruses), followed by enterovirus A71 and enterovirus B80 (1 each, 4.3%). Phylogenetically, the 9 E30 strains sequenced in our study belonged to 2 distinct genogroups, V and VIIb, and showed close relationship with E30 strains circulating in Russia and elsewhere in Asia, including China (Figure 4).
In additional to enterovirus sequences, a gemycircularvirus genome was obtained from a 12-year-old boy. Phylogenetic analysis revealed that this gemycircularvirus strain was closely related to a gemycircularvirus species previously found in CSF sample from a patient with a CNS infection of unknown origin in Sri Lanka (12); the level of amino acid identities between the 2 strains were 98.79% for replication-coding sequences and 99.3% for capsid protein–coding sequences.
We describe a viral mNGS investigation characterizing the human virome in CSF of 204 patients in Vietnam with suspected CNS infection of unknown origin. We successfully detected 4 human viral pathogens (enteroviruses, HIV, HBV, and MCV) and 1 virus species (gemycircularvirus) of unknown tropism and pathogenicity in a total of 30 (14.7%) patients. Most patients therefore remained without a known etiology, underscoring the ongoing challenge in identifying a plausible viral pathogen in CSF of patients with CNS infections.
Enteroviruses were the most common viruses, found in 11.3% (23/204) of all analyzed patients (Figure 2), most of whom were children and young adults. This age distribution of enterovirus-infected patients is consistent with observational data from a previous report from Vietnam (6), although the median age was slightly higher compared with data from other countries (13,14). Geographically, all the enterovirus-infected patients were admitted to hospitals from central and highland Vietnam, and none was from southern Vietnam. The underlying mechanism determining this observed spatial pattern of enterovirus-positive cases in this study remains unknown. Our sampling timescale perhaps was not long enough to capture the circulation of enteroviruses in Dong Thap Province. Enteroviruses were previously reported as a leading cause of CNS infection across central and southern Vietnam (6,15,16). Collectively, our findings suggest that reverse transcription PCR (RT-PCR) testing for enteroviruses should be considered in children and young adults with CNS infections.
Of the detected enteroviruses, E30 was the most common serotype. E30 is a well-known pathogen of pediatric aseptic meningitis worldwide (17). Phylogenetically, at global scale, E30 belongs to 2 different lineages with distinct patterns of circulation and spread, 1 with a global distribution and the other with geographic restriction within Asia (17). The cocirculation of 2 E30 lineages in Vietnam suggests that E30 was imported into Vietnam on at least 2 occasions. Our analyses thus also contribute to the body of knowledge about the genetic diversity of E30 strains circulating in Vietnam.
The detection of bloodborne viruses such as HBV and HIV is unlikely to have a direct link with patients’ neurologic symptoms, although HBV has previously been reported in CSF of patients with CNS infections of unknown origin (18). The detection of HIV in CSF might have been a consequence of traumatic tap occurring during the lumbar puncture, as reflected by the high number of red blood cells in 1 of 2 HIV-positive CSF samples (data not shown). However, neuroinvasion of HIV has also been reported (19). Likewise, the pathogenic potential of a gemycircularvirus genome requires further investigation, although the detection of the gemycircularvirus genome in CSF has been reported in several papers (12,18,20). The detection of MCV and papillomavirus in CSF might result from contamination of viral skin flora during lumbar puncture.
Similar to previous reports about discrepancy between mNGS and conventional diagnostic testing (8,18,21), our observations found that most mNGS-positive results were not confirmed by subsequent viral RT-PCR assays, especially the sensitive enterovirus-specific RT-PCR with a limit of detection of ≈9 copies/reaction (22). Such results could be attributable to bleedover (also called index hopping) of indices from reads of 1 sample into reads of another sample co-sequenced on the same Illumina run (R. Sinha et al., unpub. data, https://doi.org/10.1101/125724). Applying double indexes, which was not used in our study, has been shown to substantially reduce, but not eliminate, the cross-contamination phenomenon between samples in the same run.
Our study has some limitations. First, as outlined previously, we did not employ a double unique index combination strategy per sample as part of the sequencing procedure. The well-known index hopping phenomenon possibly explains the high discrepancy between confirmatory PCR and mNGS results (21,23,24) and emphasizes the usefulness of dual indexing and including no template controls. As such, we pragmatically chose to verify our mNGS by performing specific PCR on original materials. Second, the DNase treatment step in our assay meant to reduce cellular DNA concentration in CSF might reduce the sensitivity of mNGS for the detection of DNA viruses such as herpes simplex virus (25,26). Third, some of the non–PCR-confirmed viral sequences likely originated from contamination of reagents, which is a lingering problem for mNGS (27,28).
In summary, our results emphasize that mNGS could detect a broad range of viral nucleic acids in CSF. In spite of extensive investigation, establishing the etiology in many patients with CNS infections remains a challenge. However, our findings indicate that enteroviruses are important causes of viral CNS infections in Vietnam and thus should be considered in the differential diagnosis among young patients with CNS infections.
Ms. Nguyen is a PhD student in life science at Open University, Milton Keynes, UK. Her research interests are virus discovery and evolution of emerging pathogens such as enteroviruses.
The study was funded by the Wellcome Trust of Great Britain (awards nos. WT/093724 and 106680/B/14/Z [to the Oxford University Clinical Research Unit in Vietnam], 100087/Z/12/Z [to S.B.] and 204904/Z/16/Z [to L.V.T.]), and the Royal Society (grant no. 098511/Z/12/Z [to S.B.]). X.D. and E.D. were supported by the Blood Systems Research Institute and the National Heart, Lung, and Blood Institute (grant no. R01 HL105770).
VIZIONS Consortium members: from the Oxford University Clinical Research Unit, Bach Tuan Kiet, Stephen Baker, Alessandra Berto, Maciej F. Boni, Juliet E. Bryant, Bui Duc Phu, James I. Campbell, Juan Carrique-Mas, Dang Manh Hung, Dang Thao Huong, Dang Tram Oanh, Jeremy N. Day, Dinh Van Tan, H. Rogier van Doorn, Duong An Han, Jeremy J. Farrar, Hau Thi Thu Trang, Ho Dang Trung Nghia, Hoang Bao Long, Hoang Van Duong, Huynh Thi Kim Thu, Lam Chi Cuong, Le Manh Hung, Le Thanh Phuong, Le Thi Phuc, Le Thi Phuong, Le Xuan Luat, Luu Thi Thu Ha, Ly Van Chuong, Mai Thi Phuoc Loan, Behzad Nadjm, Ngo Thanh Bao, Ngo Thi Hoa, Ngo Tri Tue, Nguyen Canh Tu, Nguyen Dac Thuan, Nguyen Dong, Nguyen Khac Chuyen, Nguyen Ngoc An, Nguyen Ngoc Vinh, Nguyen Quoc Hung, Nguyen Thanh Dung, Nguyen Thanh Minh, Nguyen Thi Binh, Nguyen Thi Hong Tham, Nguyen Thi Hong Tien, Nguyen Thi Kim Chuc, Nguyen Thi Le Ngoc, Nguyen Thi Lien Ha, Nguyen Thi Nam Lien, Nguyen Thi Ngoc Diep, Nguyen Thi Nhung, Nguyen Thi Song Chau, Nguyen Thi Yen Chi, Nguyen Thieu Trinh, Nguyen Thu Van, Nguyen Van Cuong, Nguyen Van Hung, Nguyen Van Kinh, Nguyen Van Minh Hoang, Nguyen Van My, Nguyen Van Thang, Nguyen Van Thanh, Nguyen Van Vinh Chau, Nguyen Van Xang, Pham Ha My, Pham Hong Anh, Pham Thi Minh Khoa, Pham Thi Thanh Tam, Pham Van Lao, Pham Van Minh, Phan Van Be Bay, Maia A. Rabaa, Motiur Rahman, Corinne Thompson, Guy Thwaites, Ta Thi Dieu Ngan, Tran Do Hoang Nhu, Tran Hoang Minh Chau, Tran Khanh Toan, Tran My Phuc, Tran Thi Kim Hong, Tran Thi Ngoc Dung, Tran Thi Thanh Thanh, Tran Thi Thuy Minh, Tran Thua Nguyen, Tran Tinh Hien, Trinh Quang Tri, Vo Be Hien, Vo Nhut Tai, Vo Quoc Cuong, Voong Vinh Phat, Vu Thi Lan Huong, Vu Thi Ty Hang, and Heiman Wertheim; from the Centre for Immunity, Infection, and Evolution, University Of Edinburgh, Edinburgh, Scotland, UK, Carlijn Bogaardt, Margo Chase-Topping, Al Ivens, Lu Lu, Dung Nyugen, Andrew Rambaut, Peter Simmonds, and Mark Woolhouse; from The Wellcome Trust Sanger Institute, Hinxton, UK, Matthew Cotten, Bas B. Oude Munnink, Paul Kellam, and My Vu Tra Phan; from the Laboratory of Experimental Virology, Department of Medical Microbiology, Center for Infection and Immunity Amsterdam (CINIMA), Academic Medical Center of the University of Amsterdam, Amsterdam, the Netherlands, Martin Deijs, Lia van der Hoek, Maarten F. Jebbink, and Seyed Mohammad Jazaeri Farsani; and from Metabiota, California, USA, Karen Saylors and Nathan Wolfe.
Acknowledgment
We would like to thank Le Thi Kim Thanh for her logistic support and the patients for their participation in this study.
References
- Granerod J, Tam CC, Crowcroft NS, Davies NWS, Borchert M, Thomas SL. Challenge of the unknown. A systematic review of acute encephalitis in non-outbreak situations. Neurology. 2010;75:924–32. DOIPubMedGoogle Scholar
- Feigin VL, Krishnamurthi RV, Theadom AM, Abajobir AA, Mishra SR, Ahmed MB, et al.; GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16:877–97. DOIPubMedGoogle Scholar
- World Health Organization. Neurological disorders: public health challenges. 2006 [cited 2017 Nov 11]. https://www.who.int/mental_health/publications/neurological_disorders_ph_challenges
- Rabaa MA, Tue NT, Phuc TM, Carrique-Mas J, Saylors K, Cotten M, et al. The Vietnam Initiative on Zoonotic Infections (VIZIONS): a strategic approach to studying emerging zoonotic infectious diseases. EcoHealth. 2015;12:726–35. DOIPubMedGoogle Scholar
- Ho Dang Trung N, Le Thi Phuong T, Wolbers M, Nguyen Van Minh H, Nguyen Thanh V, Van MP, et al.; VIZIONS CNS Infection Network. Aetiologies of central nervous system infection in Viet Nam: a prospective provincial hospital-based descriptive surveillance study. PLoS One. 2012;7:
e37825 . DOIPubMedGoogle Scholar - Brown JR, Bharucha T, Breuer J. Encephalitis diagnosis using metagenomics: application of next generation sequencing for undiagnosed cases. J Infect. 2018;76:225–40. DOIPubMedGoogle Scholar
- Anh NT, Hong NTT, Nhu LNT, Thanh TT, Lau C-Y, Limmathurotsakul D, et al. Viruses in Vietnamese patients presenting with community-acquired sepsis of unknown cause. J Clin Microbiol. 2019;57:e00386–19. DOIPubMedGoogle Scholar
- Aiemjoy K, Altan E, Aragie S, Fry DM, Phan TG, Deng X, et al. Viral species richness and composition in young children with loose or watery stool in Ethiopia. BMC Infect Dis. 2019;19:53. DOIPubMedGoogle Scholar
- Kroneman A, Vennema H, Deforche K, v d Avoort H, Peñaranda S, Oberste MS, et al. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol. 2011;51:121–5. DOIPubMedGoogle Scholar
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Phan TG, Mori D, Deng X, Rajidrajith S, Ranawaka U, Fan Ng TF, et al. Small viral genomes in unexplained cases of human encephalitis, diarrhea, and in untreated sewage. Virology. 2015;482:98–104. DOIPubMedGoogle Scholar
- Sun Y, Miao Z, Yan J, Gong L, Chen Y, Chen Y, et al. Sero-molecular epidemiology of enterovirus-associated encephalitis in Zhejiang Province, China, from 2014 to 2017. Int J Infect Dis. 2019;79:58–64. DOIPubMedGoogle Scholar
- Richter J, Tryfonos C, Christodoulou C. Molecular epidemiology of enteroviruses in Cyprus 2008-2017. PLoS One. 2019;14:
e0220938 . DOIPubMedGoogle Scholar - Taylor WR, Nguyen K, Nguyen D, Nguyen H, Horby P, Nguyen HL, et al. The spectrum of central nervous system infections in an adult referral hospital in Hanoi, Vietnam. PLoS One. 2012;7:
e42099 . DOIPubMedGoogle Scholar - Tan V, Thai H, Phu NH, Nghia HDT, Chuong LV, Sinh DX, et al. Viral aetiology of central nervous system infections in adults admitted to a tertiary referral hospital in southern Vietnam over 12 years. PLoS Negl Trop Dis. 2014;8:
e3127 . DOIPubMedGoogle Scholar - Lema C, Torres C, Van der Sanden S, Cisterna D, Freire MC, Gómez RM. Global phylodynamics of Echovirus 30 revealed differential behavior among viral lineages. Virology. 2019;531:79–92. DOIPubMedGoogle Scholar
- Schibler M, Brito F, Zanella MC, Zdobnov EM, Laubscher F, L’Huillier AG, et al. Viral sequences detection by high-throughput sequencing in cerebrospinal fluid of individuals with and without central nervous system disease. Genes (Basel). 2019;10:1–12. DOIPubMedGoogle Scholar
- Zayyad Z, Spudich S. Neuropathogenesis of HIV: from initial neuroinvasion to HIV-associated neurocognitive disorder (HAND). Curr HIV/AIDS Rep. 2015;12:16–24. DOIPubMedGoogle Scholar
- Zhou C, Zhang S, Gong Q, Hao A. A novel gemycircularvirus in an unexplained case of child encephalitis. Virol J. 2015;12:197. DOIPubMedGoogle Scholar
- Wilson MR, Sample HA, Zorn KC, Arevalo S, Yu G, Neuhaus J, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N Engl J Med. 2019;380:2327–40. DOIPubMedGoogle Scholar
- Thanh TT, Anh NT, Tham NT, Van HM, Sabanathan S, Qui PT, et al. Validation and utilization of an internally controlled multiplex Real-time RT-PCR assay for simultaneous detection of enteroviruses and enterovirus A71 associated with hand foot and mouth disease. Virol J. 2015;12:85. DOIPubMedGoogle Scholar
- Wilson MR, Fedewa G, Stenglein MD, Olejnik J, Rennick LJ, Nambulli S, et al. Multiplexed metagenomic deep sequencing to analyze the composition of high-priority pathogen reagents. mSystems. 2016;1:1–9. DOIPubMedGoogle Scholar
- Yang J, Yang F, Ren L, Xiong Z, Wu Z, Dong J, et al. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J Clin Microbiol. 2011;49:3463–9. DOIPubMedGoogle Scholar
- Nguyen TTH, Nguyen TA, Nguyen Thi Hoang M, Ho DTN, Le NNT. Tran tan T, et al. Performance of metagenomic next-generation sequencing for the diagnosis of viral meningoencephalitis in a resource limited setting. Open Forum Infect Dis. 2020;7:ofaa046.
- Perlejewski K, Popiel M, Laskus T, Nakamura S, Motooka D, Stokowy T, et al. Next-generation sequencing (NGS) in the identification of encephalitis-causing viruses: Unexpected detection of human herpesvirus 1 while searching for RNA pathogens. J Virol Methods. 2015;226:1–6. DOIPubMedGoogle Scholar
- Asplund M, Kjartansdóttir KR, Mollerup S, Vinner L, Fridholm H, Herrera JAR, et al. Contaminating viral sequences in high-throughput sequencing viromics: a linkage study of 700 sequencing libraries. Clin Microbiol Infect. 2019;25:1277–85. DOIPubMedGoogle Scholar
- Holmes EC. Reagent contamination in viromics: all that glitters is not gold. Clin Microbiol Infect. 2019;25:1167–8. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: December 18, 2020
1Members are listed at the end of this article.
Table of Contents – Volume 27, Number 1—January 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nguyen To Anh or Le Van Tan, Oxford University Clinical Research Unit, 764 VoVan Kiet, District 5, Ho Chi Minh City, Vietnam
Top