Volume 27, Number 12—December 2021
Dispatch
SARS-CoV-2 Variants, South Sudan, January–March 2021
Figure 2
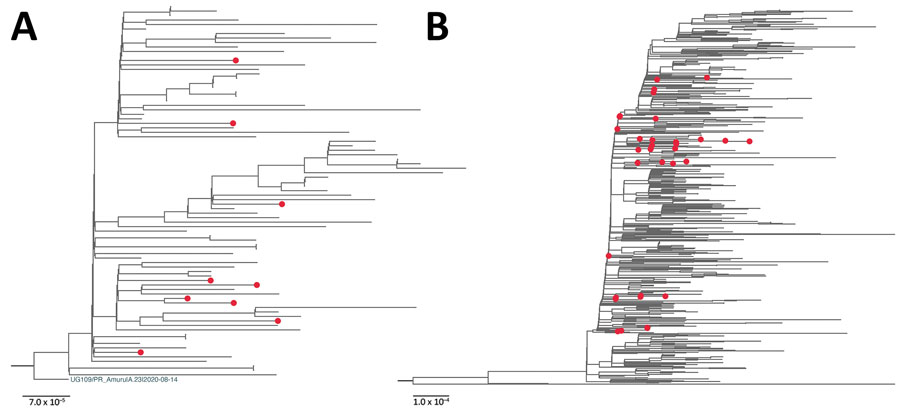
Figure 2. Maximum-likelihood phylogenetic tree of severe acute respiratory syndrome coronavirus 2 viruses from South Sudan (red dots) and reference sequences. A) Lineage A.23.1. All sequences from South Sudan were combined with a subset of all available global A.23.1 genomes, algorithmically thinned. All available global A.23.1 genomes were retrieved from GISAID (https://www.gisaid.org) and aligned, and for the first genome, all genomes closer than 5 hamming distance were removed. This process was continued until the entire set was thinned. This global, thinned A.23.1 set was combined with all South Sudan A.23.1 genomes and used to infer the A.23.1 maximum-likelihood tree. The tree was rooted with the A.23 strain (UG109/PR_Amuru|A.23|2020–08–14). B) Lineage B.1.525. The B.1.525 genome sequences were prepared in the same manner as those for A.23.1 except the hamming distance of 20. Maximum-likelihood phylogenetic trees were constructed in RaxML-NG (8) under the general time reversible plus gamma 4 plus invariate sites model as the best-fit model of substitution according to the Akaike information criterion determined by modeltestNG (9) and run for 100 pseudoreplicates and visualized using FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree). For B.1.525, the tree was midpoint rooted for clarity. Scale bar indicates nucleotide substitutions per site.
References
- Center for Systems Science and Engineering (CSSE) at Johns Hopkins University. COVID-19 dashboard. 2020 [cited 2021 Jan 24]. https://www.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6
- World Health Organization. South Sudan confirms first case of COVID-19. 2020 [cited 2021 Sep 3]. https://www.afro.who.int/news/south-sudan-confirms-first-case-covid-19
- Wiens KE, Mawien PN, Rumunu J, Slater D, Jones FK, Moheed S, et al. Seroprevalence of severe acute respiratory syndrome coronavirus 2 IgG in Juba, South Sudan, 2020. Emerg Infect Dis. 2021;27:1598–606. DOIPubMedGoogle Scholar
- World Health Organization. South Sudan joint external evaluation report 2018. 2018 [cited 2021 Sep 3]. https://www.afro.who.int/publications/south-sudan-joint-external-evaluation-report2018
- Cotten M, Bugembe DL, Kaleebu P, Phan , MVT. Alternate primers for whole-genome SARS-CoV-2 sequencing. Virus Evol. 2021; 27:veab006.
- Bugembe DL, Phan MVT, Ssewanyana I, Semanda P, Nansumba H, Dhaala B, et al. Emergence and spread of a SARS-CoV-2 lineage A variant (A.23.1) with altered spike protein in Uganda. Nat Microbiol. 2021;6:1094–101. DOIPubMedGoogle Scholar
- World Health Organization. Tracking SARS-CoV-2 variants. 2021 [cited 2021 Sep 3]. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–5. DOIPubMedGoogle Scholar
- Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2020;37:291–4. DOIPubMedGoogle Scholar
- Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021;591:293–9. DOIPubMedGoogle Scholar
- Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells.