Volume 27, Number 12—December 2021
Research Letter
SARS-CoV-2 Sequence Analysis during COVID-19 Case Surge, Liberia, 2021
Figure
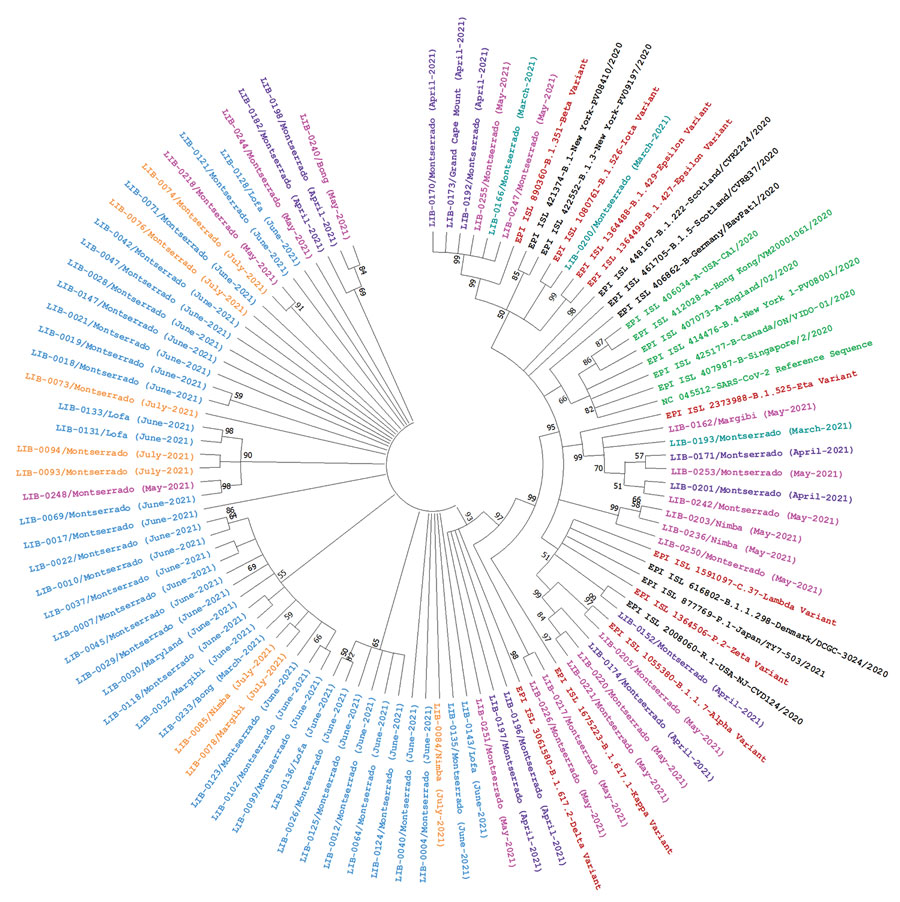
Figure. Phylogenetic analysis of 77 nasopharyngeal swab samples collected during coronavirus disease case surge, Libera, March–July 2021, and reference sequences. We created a maximum-likelihood nucleotide phylogenetic tree of the complete polyprotein coding region by using MEGA X (https://www.megasoftware.net), with a bootstrap value of 100 and and used Tamura-Nei 93 (TN93) as a substitution model with a discrete gamma distribution (+G) for evolutionary rate; the rate variation model allowed some sites to be evolutionarily invariable (+I). Numbers along the branches are bootstrap values of 100 bootstrap resamplings. Teal indicates samples collected in March 2021; purple indicates samples collected in April 2021; pink indicates samples collected in May 2021; blue indicates samples collected in June 2021; orange indicates samples collected in July 2021; brown indicates variants of concern or variants of interest; black indicates other circulating variants; green indicates severe acute respiratory syndrome coronavirus 2 reference sequence and other early parental sequences from 2020.
1These first authors contributed equally to this article.