Volume 27, Number 12—December 2021
Research Letter
SARS-CoV-2 Sequence Analysis during COVID-19 Case Surge, Liberia, 2021
Cite This Article
Citation for Media
Abstract
In June 2021, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) cases surged in Liberia. SARS-CoV-2 sequences from patients hospitalized during March–July 2021 revealed the Delta variant was in Liberia in early March and was dominant in June, irrespective of geography. Mutations and deletions suggest multiple SARS-CoV-2 Delta variant introductions.
Before May 2021, Liberia reported <10 coronavirus disease (COVID-19) cases per day among its population of ≈5 million (1). Thereafter, case numbers, hospitalizations, and deaths rapidly increased and peaked to >200 cases and 10–15 deaths per day in mid-July 2021 (Appendix Figure 1). To determine whether the rapid case surge was associated with the introduction of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern or newly emerging variants, we collected nasopharyngeal swab samples from 267 hospitalized patients countrywide during March–July 2021 for high-throughput sequencing.
We collected samples in viral transport media from Bomi, Bong, Grand Cape Mount, Lofa, Margibi, Maryland, Montserrado, and Nimba Counties (Appendix Figure 2). We noted sample collection date and site and sex and median age of patients from whom samples were obtained (Table; Appendix Table). We used Buffer AVL (QIAGEN, https://www.qiagen.com) lysis buffer to extract total nucleic acid and performed PCR by using the Triplex-CII-SARS-Cov-2 rRT PCR assay (2). We conducted further high-throughput sequencing on 89/267 (33.3%) samples that had cycle threshold values <33 (Appendix Table).
To prepare libraries, we used the Kapa Hyperplus Kit (Roche, https://www.roche.com) on first strand cDNA synthesized from 89 RNA samples (3), then we enriched for SARS-CoV-2 by using myBaits Custom RNA-Seq Kit (Daicel Arbor Biosciences, https://arborbiosci.com). We sequenced captured libraries on Nextseq 2000 or Nextseq 550 (Illumina, https://www.illumina.com), which yielded 5–8 million 220-bp reads per sample. We mapped reads to a SARS-CoV-2 reference sequence (GenBank accession no. NC_045512) to determine variants (Table; Appendix Table).
Of the 89 RNA samples, 77 (86.5%) yielded complete coding sequences with a minimum depth of ≈15× (GISAID accession nos. EPI_ISL_3547663–705, EPI_ISL_3560291, and EPI_ISL_4232122–52). Using high-throughput sequencing data, we generated consensus fasta sequences of 77 SARS-CoV-2 genomic sequences and further analyzed sequences by using Geneious R10 (https://www.geneious.com), Next-Strain (4), and GISAID (5).
Figure
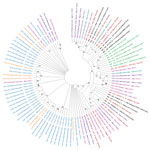
Figure. Phylogenetic analysis of 77 nasopharyngeal swab samples collected during coronavirus disease case surge, Libera, March–July 2021, and reference sequences. We created a maximum-likelihood nucleotide phylogenetic tree of the complete polyprotein...
Among 77 genomes recovered, 4 (5.2%) were Alpha variant (B.1.1.7); 6 (7.8%) were Beta variant (B.1.351); 1 (1.3%) was Iota variant (B.1.526); 6 (7.8%) were Eta variant (B.1.525); and 56 (72.7%) were Delta variant (B.1.617.2) viruses (Table). We identified Delta variant viruses in samples collected in early March and in April and May 2021, from Bong County. Delta variant viruses were co-circulating with Alpha, Beta, Eta, Iota, and other 20B variant viruses in Liberia. All 44 sequences recovered during June–July 2021 were from Delta variant viruses (Table). We used complete polyprotein coding sequences from Liberia, other representative SARS-CoV-2 sequences, and variant reference sequences to create a maximum-likelihood, nucleotide-based phylogenetic tree in MEGA X (6) (Figure).
Using reference sequence NC_045512 as a baseline, we found 3 Alpha variant–specific amino acid deletions (H69del, V70del, Y144del) in the surface glycoprotein of all Alpha variant genomes and 3 Beta variant–specific amino acid deletions (L241del, L242del, A243del) in the surface glycoprotein of all Beta variant genomes. All 56 Delta variant genomes had the 2 variant-specific amino acid deletions, F157del and R158del, and 8 of 9 other Delta variant–specific amino acid substitutions in the surface glycoprotein (T19R, G142D, E156G, L452R, T478K, D614G, P681R, and D950N). The A222V surface glycoprotein mutation was absent in only 2/56 Delta variant genomes, LIB-0226 and LIB-0217, collected from Monteserrado County in May 2021 (4). We observed another mutation in the surface glycoprotein, V367L, in 14 sequences: 1 from Bong, 2 from Margibi, 1 from Maryland, 9 from Montserrado, and 1 from Nimba. No sequences recovered from Lofa County had the V367L mutation. We noted the R724K mutation in the open reading frame 1a region of 2 sequences from Lofa, LIB-0131 and LIB-0133. LIB-0073 and LIB-0093 sequences collected from Montserrado County had 2 amino acid deletions in the open reading frame 8 region (position 120–121).
Recent surges in COVID-19 in many countries have been associated with the emergence of highly transmissible Delta variant viruses (7,8). In March 2021, the National Public Health Institute of Liberia sequenced 10 random samples from hospitalized COVID-19 patients in Monteserrado; all sequences were Alpha variant viruses (B. Shobayo, unpub. data).
A limitation of our study is the small sample sets used for analysis; nonetheless, our findings suggest that Alpha and other circulating variant viruses were replaced by Delta variant viruses countrywide in Liberia in <3 months. Mutation and phylogenetic analyses further indicate that several Delta variant strains were circulating after March 2021 and suggest multiple separate introductions.
Before June 2021, only a small percentage of the population was vaccinated in Liberia. The infections we report occurred in unvaccinated persons. The Ministry of Health, Liberia, initiated a vaccination drive in August 2021. By September, ≈130,000 persons, >2% of the population, had received a single dose of the Johnson & Johnson/Janssen vaccine (https://www.jnj.com). The COVID-19 vaccination campaign is ramping up as <30 cases/day are reported in Liberia, but the currently circulating Delta variants are a concern because they contain mutations and deletions in the surface glycoprotein that might influence vaccine efficacy (9). Liberia should continued surveillance for SARS-CoV-2 variants of concern to determine whether additional vaccination or public health measures are needed to curb severe disease and future case surges in the country.
Dr. Shobayo is public health and medical research scientist and deputy director at National Public Health Institute of Liberia, Monrovia, Liberia, and member of Partnership for Research on Infectious Diseases in Liberia. His research interests include surveillance and investigation of emerging and re-emerging viruses and their impacts on public health.
Acknowledgments
We thank Pryanka Sharma and Gilbert Smith; Melvin Johnson, John Fayiah and Julie Blie and all the members of Partnership for Research on Vaccines and Infectious Diseases in Liberia; Julius Teahton, Francis Jaryan, John Dogba, Fahn Taweh, and Joseph Tahyor; and National Public Health Institute of Liberia research and lab team members for their support in collection and transport of samples.
Financial support was provided by by the Skoll Foundation for the Global Alliance for Pandemic Prevention (GAPP grant no. 20-45017), the Tong Tsung and Wei Fong Chao Foundation (grant no. GT007457), and the Chau Hoi Sheun Foundation (grant no. GT007457).
The National Institute of Allergy and Infectious Diseases, National Institutes of Health, also participated in study design, analysis and interpretation of data, writing of the report, and decision to submit the article for publication.
References
- Worldometer. COVID-19 coronavirs disease data, Liberia [cited 2021 Sep 26]. https://www.worldometers.info/coronavirus/country/liberia
- US Food and Drug Administration. Accelerated emergency use authorization (EUA) summary the Triplex CII-SARS-CoV-2 rRT-PCR test updated 9/14/2020 [cited 2021 Sep 15]. https://www.fda.gov/media/137983/download
- Mishra N, Ng TFF, Marine RL, Jain K, Ng J, Thakkar R, et al. Antibodies to enteroviruses in cerebrospinal fluid of patients with acute flaccid myelitis. MBio. 2019;10:e01903–19. DOIPubMedGoogle Scholar
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. DOIPubMedGoogle Scholar
- Lopez Bernal J, Andrews N, Gower C, Gallagher E, Simmons R, Thelwall S, et al. Effectiveness of Covid-19 vaccines against the B.1.617.2 (Delta) variant. N Engl J Med. 2021;385:585–94. DOIPubMedGoogle Scholar
- Alizon S, Haim-Boukobza S, Foulongne V, Verdurme L, Trombert-Paolantoni S, Lecorche E, et al. Rapid spread of the SARS-CoV-2 Delta variant in some French regions, June 2021. Euro Surveill. 2021;26:
2100573 . DOIPubMedGoogle Scholar - Creech CB, Walker SC, Samuels RJ. SARS-CoV-2 vaccines. JAMA. 2021;325:1318–20. DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleOriginal Publication Date: October 27, 2021
1These first authors contributed equally to this article.
Table of Contents – Volume 27, Number 12—December 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nischay Mishra, Center for Infection and Immunity, Columbia University, 722 W 168th St, New York, NY 10032, USA
Top