Volume 31, Number 8—August 2025
Research
Rapid Emergence and Evolution of SARS-CoV-2 Intrahost Variants among COVID-19 Patients with Prolonged Infections, Singapore
Cite This Article
Citation for Media
Abstract
The evolution and spread of SARS-CoV-2 variants have driven successive waves of global COVID-19 outbreaks, yet the longitudinal dynamics of intrahost variation within the same patient remain less clear. We conducted a longitudinal cohort study by deep sequencing 198 swab samples collected from COVID-19 patients with varying infection durations. Our analysis showed that prolonged infections enhanced viral genomic diversity, leading to emergence of co-occurring variants that maintained high (>20%) frequency and became dominant in virus populations. We observed heterogeneous intrahost dynamics among individual patients, 2 of whom exhibited a minor variant of the spike D614G substitution over the course of infection. The increase in intrahost variants strongly correlated with prolonged infections, highlighting the complex interplay between viral diversity and host factors. This study revealed the intricate evolutionary mechanisms driving the emergence of de novo variants and lineage dominance, which could inform development of effective vaccine candidates and strategies to protect public health.
The COVID-19 pandemic, caused by the zoonotic SARS-CoV-2 virus, led to an unprecedented global crisis in the 21st Century. The application of advanced sequencing technologies enabled rapid identification of emerging de novo SARS-CoV-2 variants and helped elucidate how prevailing lineages were arising and spreading. Singapore was among the first countries outside China to implement rigorous COVID-19 surveillance. During the early period of the SARS-CoV-2 outbreak, from late January to early March 2020, viruses from multiple patients in Singapore exhibited a long, 382-nt deletion mutation in the open reading frame (ORF) regions ORF7b and ORF8 (1) that was later eliminated in the population, possibly because of the reduction in case counts resulting from the country’s effective control measures (2). ORF8 deletions of varying lengths have repeatedly reemerged in subsequent major variants, including Alpha, Delta, and Omicron XBB.1 (3–6).
Studies investigating the intrahost dynamics of SARS-CoV-2 virus have demonstrated that intrahost single-nucleotide variants (iSNVs) are associated with virus shredding (7), transmission bottlenecks (8,9), purifying selection (10), immunosuppression (11), and vaccinations (12). Growing attention has been directed toward determining the complexity of viral evolution during persistent infections within hosts (13–15; M. Ghafari et al., unpub. data, https://doi.org/10.1101/2024.06.21.24309297; N. Rutsinsky et al., unpub. data, https://doi.org/10.1101/2024.11.23.624482). However, the intrahost evolutionary dynamics of SARS-CoV-2 in Singapore remain largely uncharacterized. We investigated the longitudinal intrahost variation of SARS-CoV-2 in patients with varying durations of infection during early 2020.
Sample Collection
During March–May 2020, we collected a total of 198 nasopharyngeal swab samples from 20 adult hospitalized COVID-19 patients at Singapore General Hospital (SGH). Epidemiologic and clinical data included age, sex, height, weight, body mass index, underlying conditions, intensive care unit (ICU) admission, infection duration, leukocyte count, C-reactive protein (CRP) count, and remdesivir treatment.
RNA Extraction and Next-Generation Sequencing
We extracted viral RNA from swab samples and tested for the SARS-CoV-2 RNA-dependent RNA polymerase gene, as previously described (16). We generated complete SARS-CoV-2 genomes via next-generation sequencing. We conducted library preparation by using the Illumina RNA Prep Enrichment Kit (https://www.illumina.com) and performed viral enrichment by using Respiratory Virus Oligo Panel (Illumina), following manufacturer protocols. We quantified libraries by using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, https://www.thermofisher.com) and quality-checked by using 2100 Bioanalyzer (Agilent Technologies, https://www.agilent.com). We ran pooled libraries on an Illumina MiSeq platform at 2 × 250 bp. We used Trimmomatic version 0.39 (17) to quality-trim reads using a minimum read quality of 20, leading/trailing quality of 10, and a minimum length of 50. For samples collected on the first day of swab sampling, we mapped trimmed paired reads to the wild-type SARS-CoV-2 reference genome (GenBank accession no. NC_045512.2) using Burrow-Wheeler Aligner–Maximal Exact Match (18) with UGENE version 42 (19). We used Pangolin version 4.3.1 (20) to assign Pango lineages to SARS-CoV-2 genomes from patients (GISAID accession nos. EPI_ISL_19591944–57).
iSNV Analyses
To investigate within-host evolutionary dynamics of SARS-CoV-2, we used daily nasopharyngeal swab specimens collected from the 20 participants hospitalized at SGH over the course of infection, spanning up to 40 days. We deep sequenced all 198 samples, yielding 92 complete genomes from serial timepoints (Table 1). We used SAMtools (21) to identified iSNVs and generate mpileup files, then performed variant calling by using VarScan version 2.3.4 (22).
We applied rigorous quality control steps to reduce sequencing errors. First, we trimmed and filtered reads with a minimum Phred score >30. We required variants to have sequencing depth of 200–60,000 reads, a p value of <0.01, variant read depth >10×, and genome coverage >95%. Then we used the strand-filter parameter to remove variants detected predominantly on either the forward or reverse strand but not both. To minimize false-positive results and exclude potentially fixed variants, we only retained variants with frequencies of 5%–95%, following widely used minor allele frequency cutoffs (13,23,24). That threshold is well above the reported error rates for next-generation sequencing platforms, ensuring reliable variant detection (25). For samples collected on the first day of hospitalization, we used SnpEff (26) to perform variant annotation on the basis of the wild-type reference genome (7,8,27,28). For longitudinal samples, we based annotations on the reference genome of the first confirmed Singapore case (BetaCoV/Singapore/2/2020; GISAID accession no. EPI_ISL_406973) that differs from the wild-type reference genome by a single nucleotide. We used MAFFT (https://mafft.cbrc.jp) to conducted genome alignments in Geneious Prime version 2022.1.1 (https://www.geneious.com), then manually refined.
We identified iSNVs representing subconsensus genetic diversity on the basis of nucleotide composition at each genomic position (27,29) (Appendix 1 Table 1). We found iSNV counts and frequencies were consistent when we used either the wild-type or BetaCoV/Singapore/2/2020 reference genomes. We visualized iSNV frequencies and distributions by using the ggplot2 package (https://github.com/tidyverse/ggplot2) and custom scripts in R (The R Project for Statistical Computing, https://www.r-project.org). We used the ComplexHeatmap package (30) in R to display high (>20%) frequency iSNVs as heatmaps. To assess variation of iSNV counts and frequencies over the course of infection, we stratified patients by illness duration into acute (<7 days) and prolonged (>8 days) groups. That cutoff reflects earlier studies indicating that mild or moderate COVID-19 cases typically resolve within a week, but severe cases exhibit extended viral shedding (31–34). For each patient, we quantified the number of synonymous, nonsynonymous, and nonsense (stop) variants. We normalized iSNV counts per gene by length (kb). We visualized normalized values across all sampling days per patient as bar plots, indicating relative proportions of synonymous and nonsynonymous variants.
Correlation and Linear Regression Analyses
We used the corrplot package version 0.92 in R (https://CRAN.R-project.org/package=corrplot) to calculate Pearson correlation coefficients (r) for assessing associations between iSNV counts and 11 clinical variables and considered p<0.05 statistically significant. We defined iSNV counts as the number of unique genomic positions with a variant detected in >1 sample per patient. We classified correlation strength as very strong (r>0.7), strong (r = 0.5–0.7), moderate (r = 0.3–0.5), or weak (r<0.3). We further tested associations between iSNV counts and clinical parameters by using a negative binomial regression model with a log-link function in the MASS package (35) in R. We performed Wilcoxon tests to compare factors between 2 groups. We used the Benjamini-Hochberg method to correct all p values for false discovery rate.
Ethics Considerations
This study was approved by the SingHealth Centralized Institutional Review Board (CIRB reference no. 2018/3045) and the National University of Singapore (NUS) Institutional Review Board (NUS-IRB reference code 2022-320). Written informed consent was obtained from all participants. All recruited COVID-19 patients were hospitalized during the early phase of the pandemic, isolated in negative pressure rooms, and discharged only after 2 consecutive negative quantitative PCR (qPCR) tests. All samples were de-identified and processed under Biosafety Level 3 conditions.
Clinical Characteristics of Hospitalized COVID-19 Patients
The 20 enrolled patients ranged in age from 21 to 70 (median 38 + 15.4) years, and body mass index ranged from 14.7 to 31.8 (median 25.8 + 5.0) kg/m2 (Tables 1, 2; Appendix 2 Figure 1). Hospital stays varied from 3 to 40 (median 7 + 10.2) days. Five patients (P2, P3, P5, P17, and P20) received remdesivir treatment. Four patients (P3, P4, P7, and P20) had underlying conditions, including hypertension, and experienced SARS-CoV-2 infections lasting 16 to 40 days (Table 1).
iSNVs in Longitudinal SARS-CoV-2 Samples
Figure 1
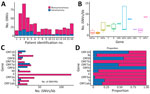
Figure 1. Distribution of iSNVs among patients in study of rapid emergence and evolution of SARS-CoV-2 intrahost variants among COVID-19 patients with prolonged infections, Singapore. A) Total number of iSNV detected in...
We analyzed subconsensus de novo iSNVs in longitudinal samples from 16 COVID-19 patients. Of 198 sequenced samples, only 92 samples had sequencing depths of 200–62,000 reads, which we included for intrahost analysis. We excluded samples from 4 patients because reads were <200 or had inadequate coverage. Among the 16 included patients, we detected 4–108 iSNVs per patient at frequencies of 5%–95% (Appendix 1 Table 2) and more nonsynonymous than synonymous mutations (Figure 1, panel A). Two patients (P2, hospitalized for 30 days, and P3, hospitalized for 40 days) exhibited higher (>70) variant counts than other patients (Table 1; Figure 1, panel A).
Unique iSNVs were unevenly distributed across the genome. ORF7b and ORF10 exhibited moderately higher iSNVs per kilobase (Figure 1, panel B), and ORF1ab harbored the highest (n = 360) number of iSNVs compared with other gene regions (n = 4–60) (Appendix 1 Table 3). Within ORF1ab, nonsynonymous (n = 261) mutations exceeded synonymous (n = 61) mutations (Appendix 1 Table 4). Nonsynonymous mutations represented >50% of all variants in most genes, except for ORF6, ORF8, and ORF10 (Figure 1, panels C, D, Appendix 1 Table 4).
Temporal Intrahost Dynamics of SARS-CoV-2 across Patients
Figure 2

Figure 2. Variant heatmaps from individual patients in study of rapid emergence and evolution of SARS-CoV-2 intrahost variants among COVID-19 patients with prolonged infections, Singapore. A) Patient 1, infected with B6.6 lineage;...
To assess the prevalence and distribution of de novo variants across SARS-CoV-2 genomes, we combined iSNV data from all longitudinal samples of 16 patients (Appendix 1 Table 1). Frequency plots revealed numerous minor variants at both low (5%–10%) and mid (10%–50%) frequencies and a notable decrease in iSNV count at >50% frequency (Appendix 2 Figure 2). We detected 9 high-frequency (>70%) variants, none of which were shared between patients. Conversely, we observed shared iSNVs in more than half the patients, and >11 shared variants detected at frequencies of 40%–70% (Appendix 2 Figure 2, panels A, B). For lower-frequency (5%–10%) variants, most were unique to individual patients, but a few were shared among multiple patients, including A7507C (ORF1a: K2414N), G10481A (ORF1a: G3406S), T15071A (ORF1b: L535I), T17190C (ORF1b: V1241A), T18402A (ORF1b: L1645Q), A20079T (ORF1b: H2204L), A21949C (spike: K129N), T23652C (spike: M697T), and A26433C (envelope: K63N) (Appendix 2 Figure 2, panel C). The K129N residues were in the N-terminal domain and the M697T residues were in the S2 subunit of the spike protein.
We observed a diverse array of iSNVs and substantial interpatient variability in both number and frequency (Figure 2; Appendix 2 Figures 3–6). Several patients, including P1, P8, P9, P13, P14, and P15, primarily harbored low-frequency (5%–20%) variants (Figure 2; Appendix 1 Table 1; Appendix 2 Figure 3). P1 exhibited more variants on day 1, most of which disappeared by day 2. That patient also harbored a unique spike substitution, A706S (Appendix 2 Figure 3), within the S2 subunit and had a short hospital stay of 5 days. By comparison, P5, who was older (>60 years of age) and hospitalized for 14 days, displayed a higher number of variants, particularly in the ORF1ab region, which appeared sporadically throughout infection (Figure 2; Appendix 2 Figure 3). That patient also carried a unique spike substitution at F823L. Patients with hospital stays >7 days, such as P2, P3, P4, P5, and P16, acquired more low-frequency variants (Figure 2; Appendix 2 Figures 3–6). Of note, P4 harbored a unique spike mutation at A397S within the receptor-binding domain of the spike protein as late as day 29 (Appendix 2 Figure 6), and P16 acquired a mutation, H1271Y, on day 8. In most patients, although some variants persisted, most either disappeared or appeared intermittently during infection.
Figure 3

Figure 3. Temporal dynamics of 76 high-frequency iSNVs in study of rapid emergence and evolution of SARS-CoV-2 intrahost variants among COVID-19 patients with prolonged infections, Singapore. Colored closed circles represent synonymous variants;...
During April–May 2020, we identified 76 variants with frequencies >20% in >1 sample (Figure 3). Because all patients were isolated, most variants likely emerged independently at specific time points. However, only 13 variants persisted during the early pandemic phase (Figure 3). Those variants included dual mutations at C6310A (nonstructural protein [NSP] 3: S1197R) and C6312A (NSP3: T1198K); co-occurrence in NSP3 has been associated with increased infection severity (34). Other persistent nonsynonymous variants included C8730T (NSP4: S59F), G11083T (NSP6: L37F), A12413C (NSP8: N108H), C19524T (NSP14: S495L), A23403G (spike: D614G), G25429T (ORF3a: V13L), and C28311T (N: P13L), suggesting those mutations were independently fixed. Among those mutations, the prominent spike D614G variant at nucleotide position 23403 might have emerged in multiple patients and coincided with S1197R (position 6310) and T1198K (position 6312), indicating a potential fitness advantage. The P13L mutation (position 28311) in the N gene has also been linked to reduced ICU admission and lower risk for death (36). Together, those findings highlight the emergence of diverse de novo synonymous and nonsynonymous variants in COVID-19 patients during the early phase of the pandemic.
Figure 4
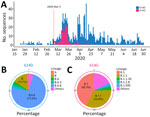
Figure 4. Evolutionary landscape in study of rapid emergence and evolution of SARS-CoV-2 intrahost variants among COVID-19 patients with prolonged infections, Singapore. A) Number of SARS-CoV-2 cases carrying the spike 614D or...
To assess the local prevalence of the spike D614G mutation, we analyzed all available SARS-CoV-2 genomes from Singapore in 2020. The G variant of S614 was detected on March 5, 2020, and its prevalence increased substantially by mid-March (Figure 4, panel A). The 614G mutation was detected in several sublineages, predominantly in B.1 (42.3%) and B.1.1 (32.9%), and the 614D variant was predominant (73.4%) in the B.6.6 lineage (Figure 4, panels B, C; Appendix 1 Table 5).
Differential Landscape of Intrahost Evolution between SARS-CoV-2 B.1 and B.6 Lineages
Figure 5

Figure 5. Comparative analysis of variants among lineages and infection durations in study of rapid emergence and evolution of SARS-CoV-2 iSNVs among COVID-19 patients with prolonged infections, Singapore. A, B) Variant frequency...
To investigate differences in intrahost evolution, we compared iSNV distributions in patients infected with B.1 or B.6/B.6.6 lineage viruses. The B.1 lineage exhibited fewer minor variants (iSNVs = 71) at 5%–20% frequency (Figure 5, panel A), whereas B.6/B.6.6 showed a marked increase (iSNVs = 185) (Figure 5, panel B). B.1 lineage also had fewer mid- to high-frequency (>20%) variants (n = 31) compared with B.6 (n = 60), although each lineage displayed a diverse set of shared high-frequency iSNVs.
In the B.1 lineage, several variants were shared among patients, including those at nucleotide positions 3037 (NSP3: F106F), 5434 (NSP3: G905G), 7507 (NSP3: K1596N), 14408 (NSP12: L323L), 15071 (NSP12: L544I), 18703 (NSP14: Q222H), 23403 (S: D614G), 20079 (NSP15: H153L), 21949 (spike: K129N), and 27750 (ORF7a: K119K) (Figure 5, panel A). In contrast, B.6/B.6.6 exhibited more low- to high-frequency iSNVs (Figure 5, panel B). However, we found only a few unique high-frequency (>20%) variants in 5 patients infected with B.6/B.6.6, including mutations at 6310 (NSP3: S1197R), 6312 (NSP3: T1198K), 11083 (NSP6: L37F), 19524 (NSP14: S495L), and 28311 (N: P13L). Spike D614G was observed at lower frequencies in B.6 patients compared with B.1.1 patients. Of note, 3 patients (P2, P3, and P4) acquired the S:D614G mutation during acute or postacute infection: P2 on day 1, P3 on day 3, and P4 as late as day 18 (Appendix 2 Figures 4–6). That time to acquisition suggests high-frequency variants might emerge over the course of infection, as in P3 and P4, who had B.6.6 lineage (Appendix 2 Figures 5, 6), but other variants might appear early, as in P16, who had B.1.1 lineage (Figure 2; Appendix 2 Figure 3).
Prolonged SARS-CoV-2 Infection and Increasing Intrahost Genetic Variability
We next compared de novo iSNVs in patients with infections <7 days versus those with 8–40 days of active infection. Patients with prolonged infections yielded more (n = 223) iSNVs across the genome than those with shorter infections (n = 93 iSNVs) (Figure 5, panels C, D). That difference was more pronounced in variants with >20% frequency (69 vs. 15). Among patients with shorter infections, most variants were at low (5%–20%) frequencies, and certain sites, such as 4329 (NSP3: I537T), 7507 (NSP3: K1596N), 17190 (NSP13: V318A), and 27750 (ORF7a: K119K), occurred sporadically. In contrast, prolonged infections exhibited 69 high-frequency (20%–80%) variants, although the fluctuation among those variants should be interpreted with caution. Notable nonsynonymous substitutions included D614G (S), S1197R and T1198K (NSP3), L37F (NSP6), V13L (ORF3a), and P13L (nucleocapsid [N]). To explore intrahost diversity during prolonged (>8 days) infection, we analyzed iSNVs during acute (<7 days) and nonacute phases. Many (n = 133) iSNVs emerged within 7 days, and most persisted beyond day 8 of infection (Appendix 2 Figure 7). Of note, patients with prolonged infections exhibited more iSNVs during the first week than those with shorter illness durations (Figure 5, panel C; Appendix 2 Figure 7).
Figure 6

Figure 6. Temporal evolution of iSNVs in study of rapid emergence and evolution of SARS-CoV-2 intrahost variants among COVID-19 patients with prolonged infections, Singapore. The dot plots illustrate iSNVs detected over time...
We further examined intrahost SARS-CoV-2 evolution in individual patients. Most patients had numerous low-frequency iSNVs on day 1 (Figure 6; Appendix 2 Figures 8–10). We observed distinct patterns across patients: P6 (7-day hospitalization) showed low-frequency variants on days 2 and 3 and had few nonsynonymous variants (e.g., at nt position 12413) that were >25% by day 5 (Figure 6, panel A). P2 (13-day hospitalization) exhibited more iSNVs, many of which disappeared by day 8 (Figure 6, panel B). Both patients were infected with B.6.6, but P2 was older (48 years of age) and treated with remdesivir and P6 (28 years of age) was not treated (Table 1).
Two patients experienced prolonged infections; P4 had a 30-day infection, and P3 had a 40-day infection. P4 displayed several high-frequency nonsynonymous variants at positions 11071 and 11083 as early as day 1 (Figure 6, panel C), suggesting founder variants were present. In contrast, P3 showed many low-frequency iSNVs throughout infection, and only a few persisted beyond 3 weeks (Figure 6, panel D). Both patients were infected with lineage B.6.6. Specifically, in P3, the spike D614G variant fluctuated in frequency (Figure 6, panel D). It first appeared at 7% on day 3 (April 10, 2020), remained <18.2% for over a week, and then rose to 60.4% by day 15 (April 22, 2020) (Appendix 2 Figure 4). In contrast, patients with shorter (<7 days) infections (P1 and P7–P15) exhibited fewer iSNVs and limited frequency variation (Appendix 2 Figures 9, 10). Those findings highlight the variability in intrahost variant abundance and dynamics among patients.
Correlation between iSNV Counts and Clinical Variables
Figure 7
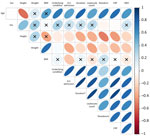
Figure 7. Correlation between iSNVs and clinical parameters in study of rapid emergence and evolution of SARS-CoV-2 intrahost variants among COVID-19 patients with prolonged infections, Singapore. Correlation matrix illustrates the relationships between...
Finally, we assessed Pearson correlations between iSNV counts and 11 clinical variables. We observed strong positive correlations with underlying conditions (r = 0.55), ICU admission (r = 0.80), infection duration (r = 0.78), remdesivir treatment (r = 0.81), leukocyte count (r = 0.66), and CRP (r = 0.78) (Table 3; Figure 7). Those variables also demonstrated strong intercorrelations, suggesting collinearity. Regression analysis further confirmed a statistically significant association between iSNV count and infection duration (p = 0.004) (Appendix 1 Table 6; Appendix 2 Figure 11). We observed no statistically significant differences between B.1 and B.6 lineages when comparing patient age or iSNV counts (Appendix 2 Figure 12). Collectively, those findings suggest host factors and treatment interventions influence the emergence of intrahost variants and contribute to viral genomic diversity.
As with most RNA viruses, SARS-CoV-2 undergoes rapid mutations and continuously generates de novo genetic variants, seeding sequential epidemics worldwide. In this study, we uncovered longitudinal intrahost dynamics of SARS-CoV-2 among hospitalized patients during the early months of the pandemic. Genomic analysis revealed a substantial number of intrahost variants emerged at varying frequencies from the first day of virus detection onwards. The low-frequency variants likely resulted from relaxed selection of a virus transmitting in an immunologically naive population or might be indicative of adaptation to the new human host. Relaxed selection on a virus population was previously observed in the first year of pandemic influenza A(H1N1) virus circulation in 2009, before the virus was subjected to immune-driven selection either from widespread infection or vaccination (37).
Intrahost population bottlenecks and natural selection play crucial roles in eliminating nonadvantageous variants (24). Several studies have indicated that intrahost variants show evidence of positive selection within persons who have persistent infections or chronic diseases or who are immunocompromised (13,38–41). Therefore, persistent infections might serve as suitable reservoirs for harboring de novo variants that can spread into the broader community. We showed that prolonged infections played a role in contributing to the broader range of genomic diversity within hosts. We also observed differential patterns of intrahost dynamics among Pango lineages. Of note, the presence of spike D614G in 3 patients with B.6 and B.6.6 lineages suggest that mutation evolved independently. However, because of stringent quarantine controls, those COVID-19 patients remained hospitalized until they tested negative by qPCR for 2 consecutive days before being discharged, preventing further transmission of that variant.
We also demonstrated that the magnitude of intrahost diversity was positively correlated with host and clinical factors. Higher leukocyte counts and increased CRP levels also have been associated with COVID-19 severity (42,43). Persistent SARS-CoV-2 infections have been shown to lead to extended periods of ongoing replication, enabling the virus to remain infectious and evolve immune escape mechanisms within hosts (44). In addition, older populations, particularly persons >65 years of age, might have impaired immune response, which has also been shown to result in a higher risk for long COVID (45) and an increased risk for reinfection with Omicron variants (46). Antiviral treatment has been suggested to contribute to greater levels of viral intrahost diversity (47).
The ongoing evolution and transmission of SARS-CoV-2 have triggered periodic epidemic waves in many countries, driven by the sequential emergence of variants over time and geographic space. Intrahost investigations have captured the dynamic patterns of population shifts, both longitudinally and cross-sectionally. Here, we showed the role of single-nucleotide variants in contributing to the overall genetic diversity and adaptive evolution of SARS-CoV-2 lineages. Collectively, both viral and host factors play major roles in the emergence and persistence of variants, which can increase the virus’s ability to evade immune-driven and vaccine-driven antibodies, displacing older lineages and potentially seeding future outbreaks.
In conclusion, we identified shared SARS-CoV-2 variants across multiple patients and found that only a limited subset of high-frequency variants predominated and persisted throughout the course of infections. We also found that prolonged infections are positively associated with increased genetic diversity, underscoring the significant role of virus–host interactions in shaping intrahost variation and evolution. Enhanced genomic sequencing and monitoring should be prioritized for vulnerable populations, such older adults, immunocompromised persons, and persons living with chronic diseases. The data generated from this study provide crucial insights into the emergence and transmission of de novo variants and can inform the development of effective vaccine candidates and strategies for protection.
Dr. Su is an associate professor at Duke-NUS Medical School in Singapore. Her research interests focus on the evolutionary and transmission dynamics of respiratory diseases in humans and animals, involving outbreak investigations particularly on influenza viruses and coronaviruses.
Acknowledgments
We thank the anonymous reviewers and Haogao Gu for invaluable suggestions. We also thank the staff at Duke-NUS Biosafety Level 3 research facility for their support and assistance in facilitating high-containment experiments in Singapore.
This study was supported and funded by Singapore National Medical Research Council’s (NMRC) Open-Fund Large Collaborative Research Grant OF-LCG/MOH-000505-05 and by contract 75N93021C00016 from the National Institute of Allergy and Infectious Diseases, US National Institutes of Health, and Duke-NUS Signature Research Programme by the Ministry of Health, Singapore.
Author contributions: Y.C.F.S., J.G.L., and G.J.D.S. conceived and designed research. J.G.L. collected clinical samples and data. Z.Y., W.F.Y., and N.G.K. performed experiments. M.A.Z. and P.C. wrote and designed in-house scripts for figures. Y.C.F.S., M.A.Z., P.C., R.Z., W.F.Y., and J.M. analysed data. Y.C.F.S., M.A.Z., P.C., and G.J.D.S drafted and wrote the manuscript, with input from A.O.T. and A.R. All authors contributed to reviewing and editing of the manuscript.
References
- Su YCF, Anderson DE, Young BE, Linster M, Zhu F, Jayakumar J, et al. Discovery and genomic characterization of a 382-nucleotide deletion in ORF7b and ORF8 during the early evolution of SARS-CoV-2. MBio. 2020;11:e01610–20. DOIPubMedGoogle Scholar
- Lin RJ, Lee TH, Lye DC. From SARS to COVID-19: the Singapore journey. Med J Aust. 2020;212:497–502.e1. DOIPubMedGoogle Scholar
- Mazur-Panasiuk N, Rabalski L, Gromowski T, Nowicki G, Kowalski M, Wydmanski W, et al. Expansion of a SARS-CoV-2 Delta variant with an 872 nt deletion encompassing ORF7a, ORF7b and ORF8, Poland, July to August 2021. Euro Surveill. 2021;26:22. DOIPubMedGoogle Scholar
- Tang Z, Yu P, Guo Q, Chen M, Lei Y, Zhou L, et al. Clinical characteristics and host immunity responses of SARS-CoV-2 Omicron variant BA.2 with deletion of ORF7a, ORF7b and ORF8. Virol J. 2023;20:106. DOIPubMedGoogle Scholar
- Feng Y, Zhao X, Luo T, Chen Z, Yang H, Chen N, et al. Emergence of a SARS-CoV-2 Omicron subvariant BA.2.2 with a 454-nucleotide genomic deletion—Sichuan Province, China, May 10, 2022. China CDC Wkly. 2022;4:904–6. DOIPubMedGoogle Scholar
- Niemeyer D, Stenzel S, Veith T, Schroeder S, Friedmann K, Weege F, et al. SARS-CoV-2 variant Alpha has a spike-dependent replication advantage over the ancestral B.1 strain in human cells with low ACE2 expression. PLoS Biol. 2022;20:
e3001871 . DOIPubMedGoogle Scholar - Ke R, Martinez PP, Smith RL, Gibson LL, Mirza A, Conte M, et al. Daily longitudinal sampling of SARS-CoV-2 infection reveals substantial heterogeneity in infectiousness. Nat Microbiol. 2022;7:640–52. DOIPubMedGoogle Scholar
- Lythgoe KA, Hall M, Ferretti L, de Cesare M, MacIntyre-Cockett G, Trebes A, et al.; Oxford Virus Sequencing Analysis Group (OVSG); COVID-19 Genomics UK (COG-UK) Consortium. SARS-CoV-2 within-host diversity and transmission. Science. 2021;372:
eabg0821 . DOIPubMedGoogle Scholar - Valesano AL, Rumfelt KE, Dimcheff DE, Blair CN, Fitzsimmons WJ, Petrie JG, et al. Temporal dynamics of SARS-CoV-2 mutation accumulation within and across infected hosts. PLoS Pathog. 2021;17:
e1009499 . DOIPubMedGoogle Scholar - Tonkin-Hill G, Martincorena I, Amato R, Lawson ARJ, Gerstung M, Johnston I, et al.; COVID-19 Genomics UK (COG-UK) Consortium; Wellcome Sanger Institute COVID-19 Surveillance Team. Patterns of within-host genetic diversity in SARS-CoV-2. eLife. 2021;10:
e66857 . DOIPubMedGoogle Scholar - Weigang S, Fuchs J, Zimmer G, Schnepf D, Kern L, Beer J, et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat Commun. 2021;12:6405. DOIPubMedGoogle Scholar
- Khateeb D, Gabrieli T, Sofer B, Hattar A, Cordela S, Chaouat A, et al. SARS-CoV-2 variants with reduced infectivity and varied sensitivity to the BNT162b2 vaccine are developed during the course of infection. PLoS Pathog. 2022;18:
e1010242 . DOIPubMedGoogle Scholar - Li J, Du P, Yang L, Zhang J, Song C, Chen D, et al. Two-step fitness selection for intra-host variations in SARS-CoV-2. Cell Rep. 2022;38:
110205 . DOIPubMedGoogle Scholar - Kemp SA, Collier DA, Datir RP, Ferreira IATM, Gayed S, Jahun A, et al.; CITIID-NIHR BioResource COVID-19 Collaboration; COVID-19 Genomics UK (COG-UK) Consortium. SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021;592:277–82. DOIPubMedGoogle Scholar
- Voloch CM, da Silva Francisco R Jr, de Almeida LGP, Brustolini OJ, Cardoso CC, Gerber AL, et al. Intra-host evolution during SARS-CoV-2 prolonged infection. Virus Evol. 2021;7:
veab078 . DOIPubMedGoogle Scholar - Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DKW, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:
2000045 . DOIPubMedGoogle Scholar - Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. DOIPubMedGoogle Scholar
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. DOIPubMedGoogle Scholar
- Okonechnikov K, Golosova O, Fursov M; UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28:1166–7. DOIPubMedGoogle Scholar
- O’Toole Á, Scher E, Underwood A, Jackson B, Hill V, McCrone JT, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:
veab064 . DOIPubMedGoogle Scholar - Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. DOIPubMedGoogle Scholar
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. DOIPubMedGoogle Scholar
- Raglow Z, Surie D, Chappell JD, Zhu Y, Martin ET, Kwon JH, et al.; Investigating Respiratory Viruses in the Acutely Ill (IVY) Network. SARS-CoV-2 shedding and evolution in patients who were immunocompromised during the omicron period: a multicentre, prospective analysis. Lancet Microbe. 2024;5:e235–46. DOIPubMedGoogle Scholar
- Wang Y, Wang D, Zhang L, Sun W, Zhang Z, Chen W, et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 2021;13:30. DOIPubMedGoogle Scholar
- Schirmer M, D’Amore R, Ijaz UZ, Hall N, Quince C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics. 2016;17:125. DOIPubMedGoogle Scholar
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92. DOIPubMedGoogle Scholar
- Gu H, Quadeer AA, Krishnan P, Ng DYM, Chang LDJ, Liu GYZ, et al. Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals. Nat Commun. 2023;14:1793. DOIPubMedGoogle Scholar
- Gonzalez-Reiche AS, Alshammary H, Schaefer S, Patel G, Polanco J, Carreño JM, et al.; PARIS/PSP study group. Sequential intrahost evolution and onward transmission of SARS-CoV-2 variants. Nat Commun. 2023;14:3235. DOIPubMedGoogle Scholar
- Markov PV, Ghafari M, Beer M, Lythgoe K, Simmonds P, Stilianakis NI, et al. The evolution of SARS-CoV-2. Nat Rev Microbiol. 2023;21:361–79. DOIPubMedGoogle Scholar
- Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2. DOIPubMedGoogle Scholar
- Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–9. DOIPubMedGoogle Scholar
- Young BE, Ong SWX, Kalimuddin S, Low JG, Tan SY, Loh J, et al.; Singapore 2019 Novel Coronavirus Outbreak Research Team. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA. 2020;323:1488–94. DOIPubMedGoogle Scholar
- Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19:141–54. DOIPubMedGoogle Scholar
- Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol. 2022;20:270–84. DOIPubMedGoogle Scholar
- Venables WNRB. Modern applied statistics with S. 4th ed. New York: Springer; 2002.
- Alsuwairi FA, Alsaleh AN, Alsanea MS, Al-Qahtani AA, Obeid D, Almaghrabi RS, et al. Association of SARS-CoV-2 nucleocapsid protein mutations with patient demographic and clinical characteristics during the Delta and Omicron waves. Microorganisms. 2023;11:1288. DOIPubMedGoogle Scholar
- Su YCF, Bahl J, Joseph U, Butt KM, Peck HA, Koay ESC, et al. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat Commun. 2015;6:7952. DOIPubMedGoogle Scholar
- Ghafari M, Hall M, Golubchik T, Ayoubkhani D, House T, MacIntyre-Cockett G, et al.; Wellcome Sanger Institute COVID-19 Surveillance Team; COVID-19 Infection Survey Group; COVID-19 Genomics UK (COG-UK) Consortium. Prevalence of persistent SARS-CoV-2 in a large community surveillance study. Nature. 2024;626:1094–101. DOIPubMedGoogle Scholar
- Choi B, Choudhary MC, Regan J, Sparks JA, Padera RF, Qiu X, et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N Engl J Med. 2020;383:2291–3. DOIPubMedGoogle Scholar
- Chaguza C, Hahn AM, Petrone ME, Zhou S, Ferguson D, Breban MI, et al.; Yale SARS-CoV-2 Genomic Surveillance Initiative. Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection. Cell Rep Med. 2023;4:
100943 . DOIPubMedGoogle Scholar - Wagner C, Kistler KE, Perchetti GA, Baker N, Frisbie LA, Torres LM, et al. Positive selection underlies repeated knockout of ORF8 in SARS-CoV-2 evolution. Nat Commun. 2024;15:3207. DOIPubMedGoogle Scholar
- Wang G, Wu C, Zhang Q, Wu F, Yu B, Lv J, et al. C-reactive protein level may predict the risk of COVID-19 aggravation. Open Forum Infect Dis. 2020;7:
ofaa153 . DOIPubMedGoogle Scholar - Bhargava A, Fukushima EA, Levine M, Zhao W, Tanveer F, Szpunar SM, et al. Predictors for severe COVID-19 infection. Clin Infect Dis. 2020;71:1962–8. DOIPubMedGoogle Scholar
- Hettle D, Hutchings S, Muir P, Moran E; COVID-19 Genomics UK (COG-UK) consortium. Persistent SARS-CoV-2 infection in immunocompromised patients facilitates rapid viral evolution: Retrospective cohort study and literature review. Clin Infect Pract. 2022;16:
100210 . DOIPubMedGoogle Scholar - Mansell V, Hall Dykgraaf S, Kidd M, Goodyear-Smith F. Long COVID and older people. Lancet Healthy Longev. 2022;3:e849–54. DOIPubMedGoogle Scholar
- Breznik JA, Rahim A, Zhang A, Ang J, Stacey HD, Bhakta H, et al. Early Omicron infection is associated with increased reinfection risk in older adults in long-term care and retirement facilities. EClinicalMedicine. 2023;63:
102148 . DOIPubMedGoogle Scholar - Heyer A, Günther T, Robitaille A, Lütgehetmann M, Addo MM, Jarczak D, et al. Remdesivir-induced emergence of SARS-CoV2 variants in patients with prolonged infection. Cell Rep Med. 2022;3:
100735 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 31, Number 8—August 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Yvonne C.F. Su, Programme in Emerging Infectious Diseases, Duke-NUS Medical School, 8 College Rd, 169857, Singapore
Top