Volume 27, Number 2—February 2021
Research
Hepatitis C Virus Transmission Clusters in Public Health and Correctional Settings, Wisconsin, USA, 2016–20171
Figure 4
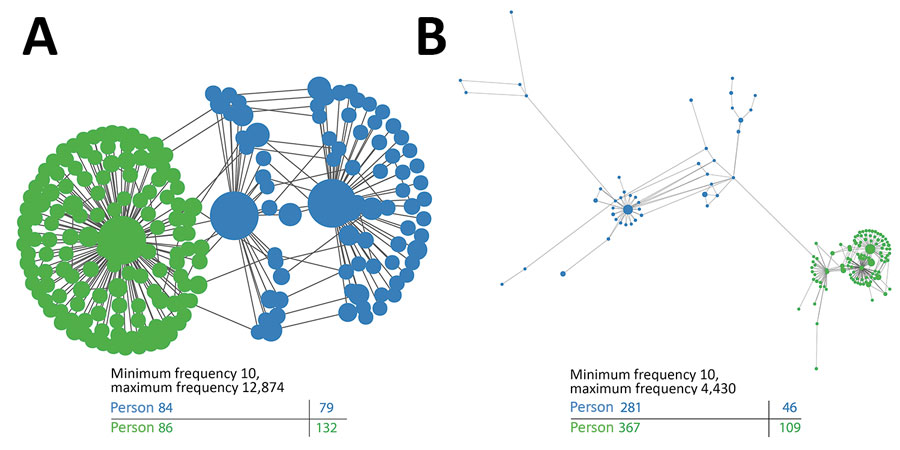
Figure 4. Intrahost genetic variation of representative transmission clusters of hepatitis C virus (HCV) among persons in public health and corrections settings, Wisconsin, USA, 2016–2017, highlighting the genetic relatedness of distinct variants. K-step network contains all possible minimum spanning trees and enables efficient visualization of genetic relatedness among all intrahost hypervariable region 1 (HVR1) variants for persons 84 and 86 (A) and persons 281 and 367 (B). Each node represents an HCV sequence. Color of the node corresponds to the sample of origin: green, found only in the first sample; blue, found only in the second sample. The node size is based on frequency of the HVR1 variant, and edge length is proportional to the modified Hamming distance (does not count positions with insertions or deletions as differences).
1The methods described in this article, along with preliminary results from the first 231 specimens analyzed, were presented in poster format at the 8th International Conference on Hepatitis Care in Substance Users; 2019 Sep 11–13; Montreal, Quebec, Canada.
2These first authors contributed equally to this article.
3These senior authors contributed equally to this article.