Volume 27, Number 6—June 2021
Research Letter
Incursion of Novel Highly Pathogenic Avian Influenza A(H5N8) Virus, the Netherlands, October 2020
Cite This Article
Citation for Media
Abstract
Highly pathogenic avian influenza A(H5N8) virus was detected in mute swans in the Netherlands during October 2020. The virus shares a common ancestor with clade 2.3.4.4b viruses detected in Egypt during 2018–2019 and has similar genetic composition. The virus is not directly related to H5N8 viruses from Europe detected in the first half of 2020.
Introduction of highly pathogenic avian influenza (HPAI) H5 clade 2.3.4.4 viruses in Europe caused substantial losses to the poultry industry during 2014–2020. Migratory waterfowl are implicated in the distribution of HPAI H5 viruses along flyways from breeding grounds in northern Russia to wintering sites in Europe (1–3). During 2016, clade 2.3.4.4b HPAI H5N8 viruses were introduced in Europe (4,5) and the Netherlands (6,7). More recent introductions of these viruses were detected in eastern Europe, Germany, and Bulgaria in the first half of 2020 (8,9).
On October 17, 2020, two mute swans (Cygnus olor) were found dead in the province of Utrecht, the Netherlands. The swans were diagnostically tested as part of the wild bird surveillance program for avian influenza virus. Swab samples from the trachea and cloaca were PCR-positive for avian influenza virus. The virus was subtyped as HPAI H5N8 and contained the hemagglutinin (HA) cleavage site sequence PLREKRRKR*GLF.
Figure
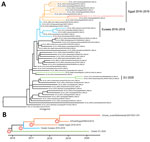
Figure. Phylogenetic analysis of the hemagglutinin (HA) segment of highly pathogenic avian influenza A(H5N8) virus, the Netherlands, October 2020. A) Optimal phylogenetic tree was generated by using the maximum-likelihood method (RAxML...
We performed full-genome sequencing as described (6) and classified the virus genetically as H5 clade 2.3.4.4b. We performed detailed phylogenetic analyses to study the origin of the novel H5N8 virus (A/mute_swan/Netherlands/20015931–001/2020, GISAID accession no. EPI591075; https://www.gisaid.org). For HA (Figure) and neuraminidase (NA) (Appendix 1 Figure 1), the closest genetic relative was isolated from a duck in Egypt during January 2019 (EPI399644; only HA/NA sequences are available). The virus also shares a common ancestor with other viruses detected in Egypt during 2018–2019 and with viruses detected in the Netherlands and Eurasia during 2016–2018. The HA and NA gene segments of the novel H5N8 virus do not cluster with the H5N8 viruses that caused widespread outbreaks in eastern Europe and Germany earlier in 2020 or with viruses detected in Bulgaria during 2020.
For the other gene segments of the novel H5N8 virus, except for the matrix (M) protein segment, clustering was also observed with H5N8 viruses that circulated in Egypt during 2018–2019 and in Eurasia during 2016–2018 (Appendix 1 Figure 1). However, the M segment clusters with HPAI H5N8 viruses isolated in Asia and Egypt in 2016–2018 but also with the viruses found in eastern Europe and Germany during 2020, which suggests that reassortment with those viruses probably occurred for the M segment. No reassortments with low pathogenicity avian influenza viruses were observed for any of the segments. The genetic distance between the novel H5N8 virus and related viruses detected in Egypt and Eurasia appears relatively large, as demonstrated by the long branch lengths in phylogenetic trees (Appendix 1 Figure 1). This finding suggests long-term, undetected circulation of the virus or that intermediate virus sequences were not available in public databases.
We performed molecular dating by using BEAST (10) to estimate the time to the most recent common ancestor (Table; Appendix 1 Figure 2). For the H5 segment, a common ancestor of the novel H5N8 virus and the Egypt 2019 virus (accession no. EPI399644) was dated to July 2018 (node 1; Appendix 1 Figure 2) and with the cluster of viruses from Egypt to approximately March 2017 (node 2; Appendix 1 Figure 2). The common ancestor for the viruses from Eurasia detected during 2016–2018 was dated to August 2016 (node 3; Appendix 1 Figure 2) and with the viruses from eastern Europe and Germany detected in 2020 to approximately December 2015 (node 4; Appendix 1 Figure 2). Similar dating of ancestral viruses was observed for other gene segments, except for M (Appendix 1 Figure 2), for which the common ancestor for the viruses from eastern Europe and Germany detected during 2020 was dated to approximately May 2016 (node A; Appendix 1 Figure 2).
Molecular dating analysis suggests that the ancestor of the novel H5N8 virus detected in the Netherlands during October 2020 has circulated in this genetic form since March 2017 and caused influenza outbreaks in Egypt during 2018–2019. The novel virus incursion is not related to viruses detected in eastern Europe, Germany, and Bulgaria earlier in 2020 but was probably associated with fall migration of wild birds to wintering sites in the Netherlands. Although no HPAI viruses or deaths were observed at wild bird breeding sites in northern Russia, HPAI H5N8 viruses were reported in southern Russia and northern Kazakhstan in September 2020. Some waterfowl species, such as Eurasian wigeon (Anas penelope), tufted duck (Aythya fuligula), and white-fronted goose (Anser albifrons), are known to migrate from these regions to the Netherlands (Dutch Centre For Field Ornithology, https://vogeltrekatlas.nl/soortzoek2.html).
The novel virus was first detected in 2 mute swans that do not migrate over long distances. However, a few days later, virus was also detected in a dead Eurasian wigeon, suggesting that this bird species might have been involved in the incursion of the virus into the Netherlands. Because sequences of the viruses detected in Russia and Kazakhstan are unknown, the relationship between these viruses and the virus detected in the Netherlands remains to be determined. During October, wild bird migration is ongoing, and millions of wild birds will reach their wintering sites in Europe in the coming months. This early detection of HPAI H5N8 virus in the Netherlands predicted a high risk for the poultry industry in Europe during the 2020–2021 winter season.
Dr Beerens is a virologist and head of the National Reference Laboratory for Avian Influenza and Newcastle Disease, Lelystad, the Netherlands. Her primary research interests are molecular virology, genetics, and virus evolution.
Acknowledgments
We thank Alex Bossers for providing excellent next-generation sequencing facilities at Wageningen Bioveterinary Research and the authors and submitting laboratories for providing sequences from the GISAID EpiFlu Database (Appendix 2).
This study was supported by the Dutch Ministry of Agriculture, Nature and Food Quality (projects WOT-01-003-087 and KB-37-003-015).
References
- Gilbert M, Xiao X, Domenech J, Lubroth J, Martin V, Slingenbergh J. Anatidae migration in the western Palearctic and spread of highly pathogenic avian influenza H5NI virus. Emerg Infect Dis. 2006;12:1650–6. DOIPubMedGoogle Scholar
- The Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science. 2016;354:213–7. DOIGoogle Scholar
- Lee DH, Sharshov K, Swayne DE, Kurskaya O, Sobolev I, Kabilov M, et al. Novel reassortant clade 2.3.4.4 avian influenza A(H5N8) virus in wild aquatic birds, Russia, 2016. Emerg Infect Dis. 2017;23:359–60. DOIPubMedGoogle Scholar
- Napp S, Majó N, Sánchez-Gónzalez R, Vergara-Alert J. Emergence and spread of highly pathogenic avian influenza A(H5N8) in Europe in 2016-2017. Transbound Emerg Dis. 2018;65:1217–26. DOIPubMedGoogle Scholar
- Lycett SJ, Pohlmann A, Staubach C, Caliendo V, Woolhouse M, Beer M, et al.; Global Consortium for H5N8 and Related Influenza Viruses. Genesis and spread of multiple reassortants during the 2016/2017 H5 avian influenza epidemic in Eurasia. Proc Natl Acad Sci U S A. 2020;117:20814–25. DOIPubMedGoogle Scholar
- Beerens N, Heutink R, Bergervoet SA, Harders F, Bossers A, Koch G. Multiple reassorted ruses as cause of highly pathogenic avian influenza A(H5N8) virus epidemic, the Netherlands, 2016. Emerg Infect Dis. 2017;23:1974–81. DOIPubMedGoogle Scholar
- Bergervoet SA, Ho CKY, Heutink R, Bossers A, Beerens N. Spread of highly pathogenic avian influenza (HPAI) H5N5 viruses in Europe in 2016. Viruses. 2019;11:
E501 . DOIPubMedGoogle Scholar - King J, Schulze C, Engelhardt A, Hlinak A, Lennermann SL, Rigbers K, et al. Novel HPAIV H5N8 reassortant (clade 2.3.4.4b) detected in Germany. Viruses. 2020;12:
E281 . DOIPubMedGoogle Scholar - Adlhoch C, Fusaro A, Kuiken T, Niqueux E, Staubach C, Terregino C, et al.; European Food Safety Authority; European Centre for Disease Prevention and Control and European Union Reference Laboratory for Avian Influenza. Avian influenza overview February - May 2020. EFSA J. 2020;18:
e06194 .PubMedGoogle Scholar - Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:
vey016 . DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleOriginal Publication Date: May 10, 2021
Table of Contents – Volume 27, Number 6—June 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nancy Beerens, Wageningen Bioveterinary Research, Division of Virology, PO Box 65, 8200 AB, Lelystad, the Netherlands
Top