Volume 27, Number 7—July 2021
Research
Fatal Human Infection with Evidence of Intrahost Variation of Eastern Equine Encephalitis Virus, Alabama, USA, 2019
Figure 1
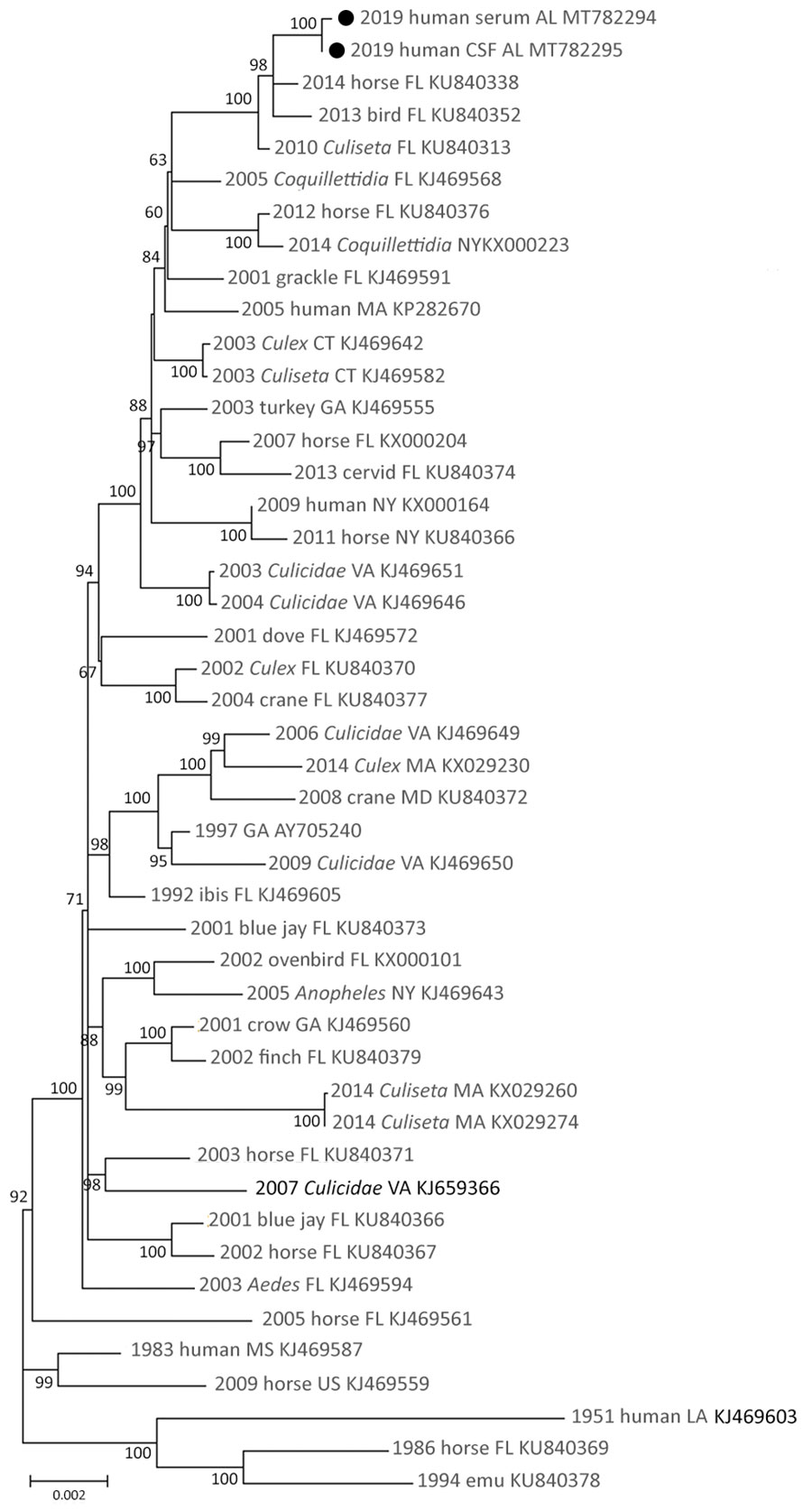
Figure 1. Maximum-likelihood phylogeny of eastern equine encephalitis virus from a woman in Alabama, USA, 2019 (solid circles), and reference sequences, based on complete coding region sequences. Nucleotide coding sequences of the full genome of eastern equine encephalitis viruses isolated during 1951–2019 were codon aligned and phylogenies inferred with general time reversible plus gamma plus proportion of invariable sites. Taxa are labeled with year of isolation, host, US state of isolation, and GenBank accession number. Branches are labeled with bootstrap support values as a percentage of 1,000 replicates. Branches with <50% bootstrap support are collapsed; only branches with >60% support are labeled. Branch lengths are drawn to scale and measured in the number of substitutions per site.