Volume 28, Number 11—November 2022
Dispatch
Cluster of Norovirus Genogroup IX Outbreaks in Long-Term Care Facilities, Utah, USA, 2021
Figure 2
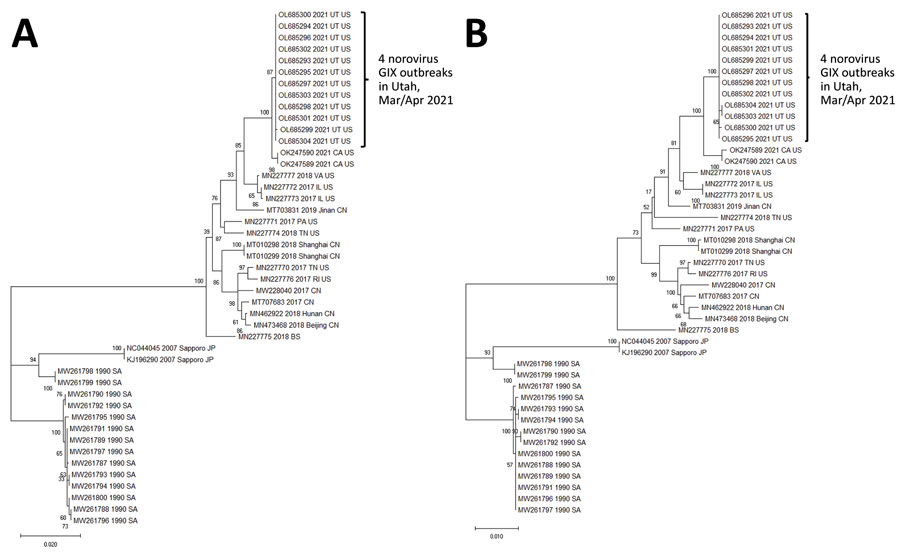
Figure 2. Phylogenetic comparisons of norovirus genes in study of cluster of norovirus genogroup IX outbreaks in long-term care facilities, Utah, USA, 2021. We generated phylogenetic trees by using the maximum-likelihood method and Tamura-Nei distance model (15). We compared nucleotide sequences of the RdRp gene (1,430 nt) (A) and major capsid gene (1,668 nt) (B) from the 12 sequences obtained from the 4 LTCF outbreaks with 33 GIX strains obtained from GenBank. The bootstrap percentages are shown next to the branches. We generated initial trees automatically by applying neighbor-joining algorithms to a matrix of pairwise distances estimated by using the maximum composite-likelihood approach and then selecting the topology with the superior log-likelihood value. We conducted evolutionary analyses by using MEGA11 software (15). Scale bars indicate nucleotide substitutions per site.
References
- Ahmed SM, Hall AJ, Robinson AE, Verhoef L, Premkumar P, Parashar UD, et al. Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. Lancet Infect Dis. 2014;14:725–30. DOIPubMedGoogle Scholar
- Wikswo ME, Kambhampati A, Shioda K, Walsh KA, Bowen A, Hall AJ; Centers for Disease Control and Prevention (CDC). Outbreaks of acute gastroenteritis transmitted by person-to-person contact, environmental contamination, and unknown modes of transmission—United States, 2009–2013. MMWR Surveill Summ. 2015;64:1–16. DOIPubMedGoogle Scholar
- Tohma K, Lepore CJ, Martinez M, Degiuseppe JI, Khamrin P, Saito M, et al. Genome-wide analyses of human noroviruses provide insights on evolutionary dynamics and evidence of coexisting viral populations evolving under recombination constraints. PLoS Pathog. 2021;17:
e1009744 . DOIPubMedGoogle Scholar - Chhabra P, de Graaf M, Parra GI, Chan MC, Green K, Martella V, et al. Updated classification of norovirus genogroups and genotypes. J Gen Virol. 2019;100:1393–406. DOIPubMedGoogle Scholar
- Cannon JL, Barclay L, Collins NR, Wikswo ME, Castro CJ, Magaña LC, et al. Genetic and epidemiologic trends of norovirus outbreaks in the United States from 2013 to 2016 demonstrated emergence of novel GII.4 recombinant viruses. J Clin Microbiol. 2017;55:2208–21. DOIPubMedGoogle Scholar
- Jin M, Wu S, Kong X, Xie H, Fu J, He Y, et al. Norovirus outbreak surveillance, China, 2016–2018. Emerg Infect Dis. 2020;26:437–45. DOIPubMedGoogle Scholar
- Kabue JP, Meader E, Hunter PR, Potgieter N. Genetic characterisation of Norovirus strains in outpatient children from rural communities of Vhembe district/South Africa, 2014-2015. J Clin Virol. 2017;94:100–6. DOIPubMedGoogle Scholar
- Sarmento SK, de Andrade JDSR, Miagostovich MP, Fumian TM. Virological and epidemiological features of norovirus infections in Brazil, 2017–2018. Viruses. 2021;13:1724. DOIPubMedGoogle Scholar
- Supadej K, Khamrin P, Kumthip K, Kochjan P, Yodmeeklin A, Ushijima H, et al. Wide variety of recombinant strains of norovirus GII in pediatric patients hospitalized with acute gastroenteritis in Thailand during 2005 to 2015. Infect Genet Evol. 2017;52:44–51. DOIPubMedGoogle Scholar
- Vega E, Barclay L, Gregoricus N, Shirley SH, Lee D, Vinjé J. Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J Clin Microbiol. 2014;52:147–55. DOIPubMedGoogle Scholar
- Chhabra P, Browne H, Huynh T, Diez-Valcarce M, Barclay L, Kosek MN, et al. Single-step RT-PCR assay for dual genotyping of GI and GII norovirus strains. J Clin Virol. 2021;134:
104689 . DOIPubMedGoogle Scholar - Tatusov RL, Chhabra P, Diez-Valcarce M, Barclay L, Cannon JL, Vinjé J. Human Calicivirus Typing tool: A web-based tool for genotyping human norovirus and sapovirus sequences. J Clin Virol. 2021;134:
104718 . DOIPubMedGoogle Scholar - Parra GI, Squires RB, Karangwa CK, Johnson JA, Lepore CJ, Sosnovtsev SV, et al. Static and evolving norovirus genotypes: implications for epidemiology and immunity. PLoS Pathog. 2017;13:
e1006136 . DOIPubMedGoogle Scholar - Wagner DD, Marine RL, Ramos E, Ng TFF, Castro CJ, Okomo-Adhiambo M, et al. VPipe: an automated bioinformatics platform for assembly and management of viral next-generation sequencing data. Microbiol Spectr. 2022;10:
e0256421 . DOIPubMedGoogle Scholar - Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38:3022–7. DOIPubMedGoogle Scholar
1These first authors contributed equally to this article.