Volume 29, Number 12—December 2023
Research Letter
Novel Ozark Orthohantavirus in Hispid Cotton Rats (Sigmodon hispidus), Arkansas, USA
Figure
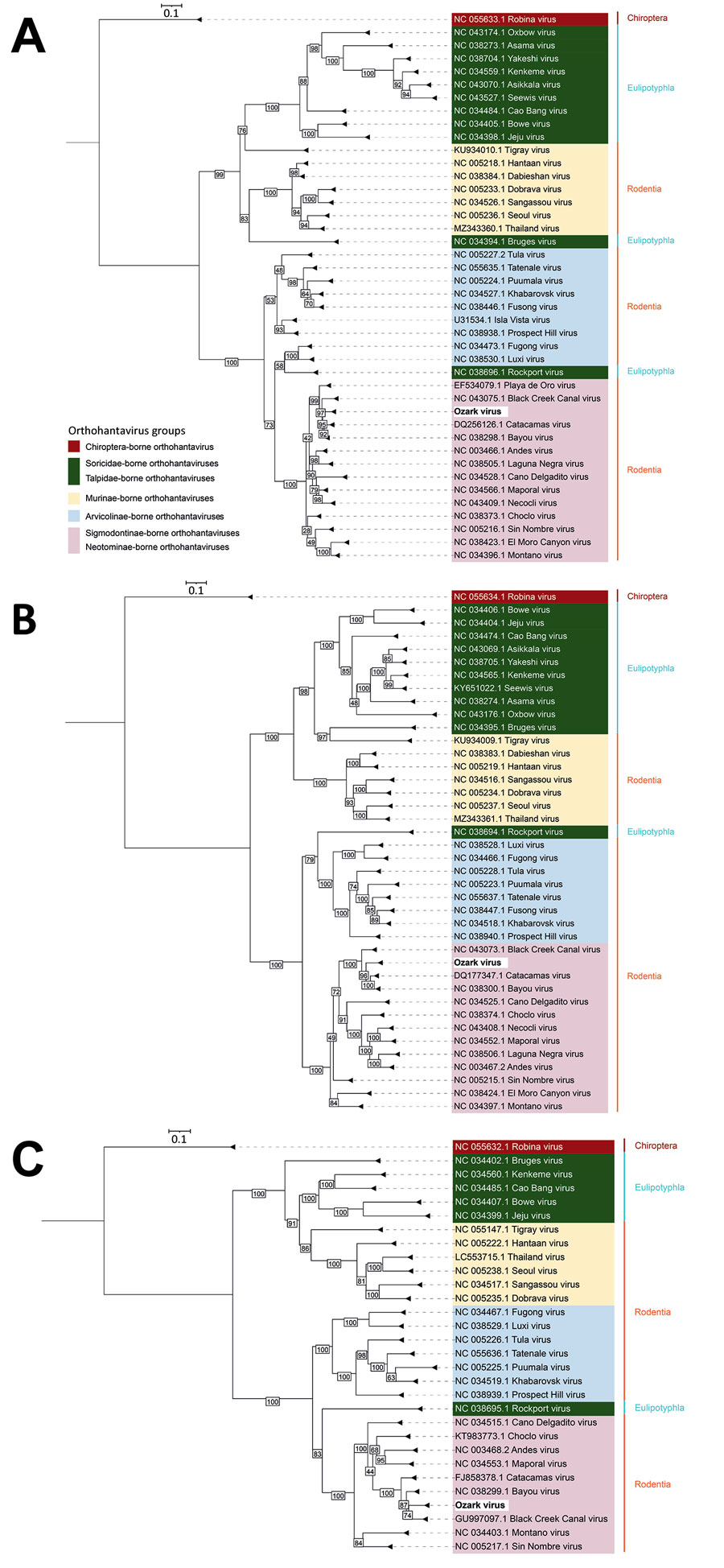
Figure. Phylogenetic analysis of novel Ozark orthohantavirus segments isolated from hispid cotton rats (Sigmodon hispidus), Arkansas, USA. Phylogenetic trees were constructed by using IQ-TREE2 (http://www.iqtree.org) for small (A), medium (B), and large (C) protein segments translated from Ozark orthohantavirus open reading frames (ORFs). ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder) was used to detect ORFs and the Expasy translate tool (https://www.expasy.org) was used to translate ORFs to amino acid sequences. Maximum-likelihood method and best-fit models Q matrix estimated for insects (Q.insect) plus proportion of invariable sites enabled plus discreet gamma model added with 4 rate categories (for small segment), Q.insect plus FreeRate model with 5 categories (for medium segment), and Q.insect plus proportion of invariable sites enabled plus invariable sites plus FreeRate model with 4 categories (for large segment) were used (http://www.iqtree.org/doc/Substitution-Models). Sequences and corresponding GenBank accession numbers are indicated for available orthohantaviruses from orders Chiroptera (bats), Eulipotyphla, and Rodentia. Orthohantaviruses from Eulipotyphla are found in families Soricidae (shrews) and Talpidae (moles); orthohantaviruses from Rodentia are found in family Muridae, subfamily Murinae (Old World mice and rats) and family Cricetidae, subfamilies Arvicolinae (voles and lemmings) and Sigmodontinae and Neotominae (both New World mice and rats). Hispid cotton rats are sigmodontine rodents. Scale bar indicates amino acid substitutions per site.
1Current affiliation: Shawnee State University, Portsmouth, Ohio, USA.