Emergence of Novel Norovirus GII.4 Variant
Preeti Chhabra
1
, Damien C. Tully
1, Janet Mans, Sandra Niendorf, Leslie Barclay, Jennifer L. Cannon, Anna M. Montmayeur, Chao-Yang Pan, Nicola Page, Rachel Williams, Helena Tutill, Sunando Roy, Cristina Celma, Stuart Beard, Michael L. Mallory, Gédéon Prince Manouana, Thirumalaisamy P. Velavan, Ayola Akim Adegnika, Peter G. Kremsner, Lisa C. Lindesmith, Stéphane Hué, Ralph S. Baric, Judith Breuer, and Jan Vinjé
Author affiliations: Centers for Disease Control and Prevention, Atlanta, Georgia, USA (P. Chhabra, L. Barclay, J.L. Cannon, A.M. Montmayeur, J. Vinjé); London School of Hygiene & Tropical Medicine, London, UK (D.C. Tully, S. Hué); University of Pretoria, Pretoria, South Africa (J. Mans, N. Page); Robert Koch Institut, Berlin, Germany (S. Niendorf); California Department of Public Health, Richmond, California, USA (C.-Y. Pan); National Institute for Communicable Diseases, Sandringham, South Africa (N. Page); UCL Great Ormond Street Institute of Child Health, London (R. Williams, H. Tutill, S. Roy, J. Breuer); UK Health Security Agency, London (C. Celma, S. Beard); University of North Carolina, Chapel Hill, North Carolina, USA (M.L. Mallory, L.C. Lindesmith, R.S. Baric); Universitätsklinikum Tübingen, Tübingen, Germany (G.P. Manouana, T.P. Velavan, A.A. Adegnika); Centre de Recherches Médicales de Lambaréné, Lambarene, Gabon (G.P. Manouana, A.A. Adegnika, P.G. Kremsner); Vietnamese-German Center for Medical Research, Hanoi, Vietnam (T.P. Velavan); Duy Tan University, Da Nang, Vietnam (T.P. Velavan); German Center for Infection Research, Tübingen (A.A. Adegnika)
Main Article
Figure 1
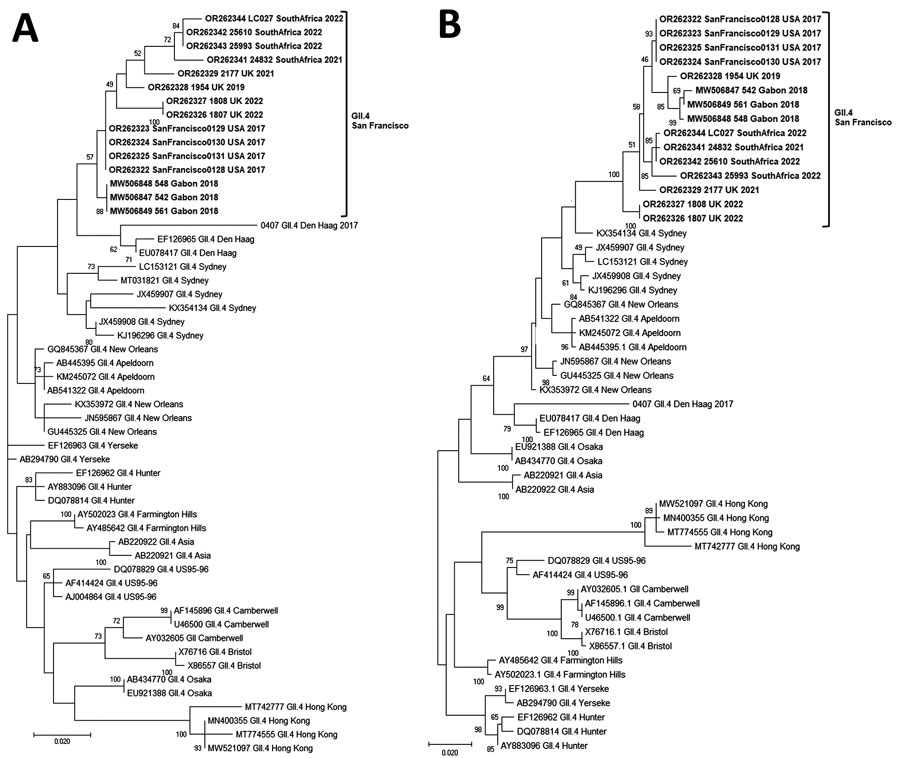
Figure 1. Phylogenetic trees of the emerging novel norovirus GII.4 strains on 3 continents. A) Common genotyping region C; 250 nt from the 5′ end of ORF2; B) complete VP1 aa sequences. Phylogenetic trees show novel GII.4 San Francisco strains and GII.4 variants, including recently identified clusters like GII.4 Hong Kong. Evolutionary analyses were conducted in MEGA X (https://www.megasoftware.net) using the maximum-likelihood method based on the Tamura-Nei model for the C region and Jones-Taylor-Thornton matrix-based model for VP1. We used a discrete gamma distribution to model evolutionary rate differences among sites; 5 categories γ parameter = 0.2174. Bootstrap (100) values are indicated at the nodes. Trees were drawn to scale. Scale bar represents nucleotide substitutions per site. ORF, open reading frame; VP1, viral protein 1.
Main Article
Page created: November 03, 2023
Page updated: December 20, 2023
Page reviewed: December 20, 2023
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.