Domestic Cat Hepadnavirus Infection in Iberian Lynxes
Georgia Diakoudi, Sabrina Castro-Scholten, Javier Caballero-Gómez, Barbara Di Martino, Federica Di Profio, Vittorio Sarchese, Francesco Pellegrini, Gianvito Lanave, Nicola Decaro, Ignacio García-Bocanegra, and Vito Martella

Author affiliation: University of Bari Aldo Moro, Bari, Italy (G. Diakoudi, F. Pellegrini, G. Lanave, N. Decaro, V. Martella); University of Córdoba, Córdoba, Spain (S. Castro-Scholten, J. Caballero-Gómez, I. García-Bocanegra); Centro de Investigación Biomédia en Red–Intermedadaes Infecciosas, Instituto de Salud Carlos III, Madrid, Spain (J. Caballero-Gómez, I. García-Bocanegra); University of Teramo, Italy (B. Di Martino, F. Di Profio, V. Sarchese); University of Veterinary Medicine, Budapest, Hungary (V. Martella)
Main Article
Figure
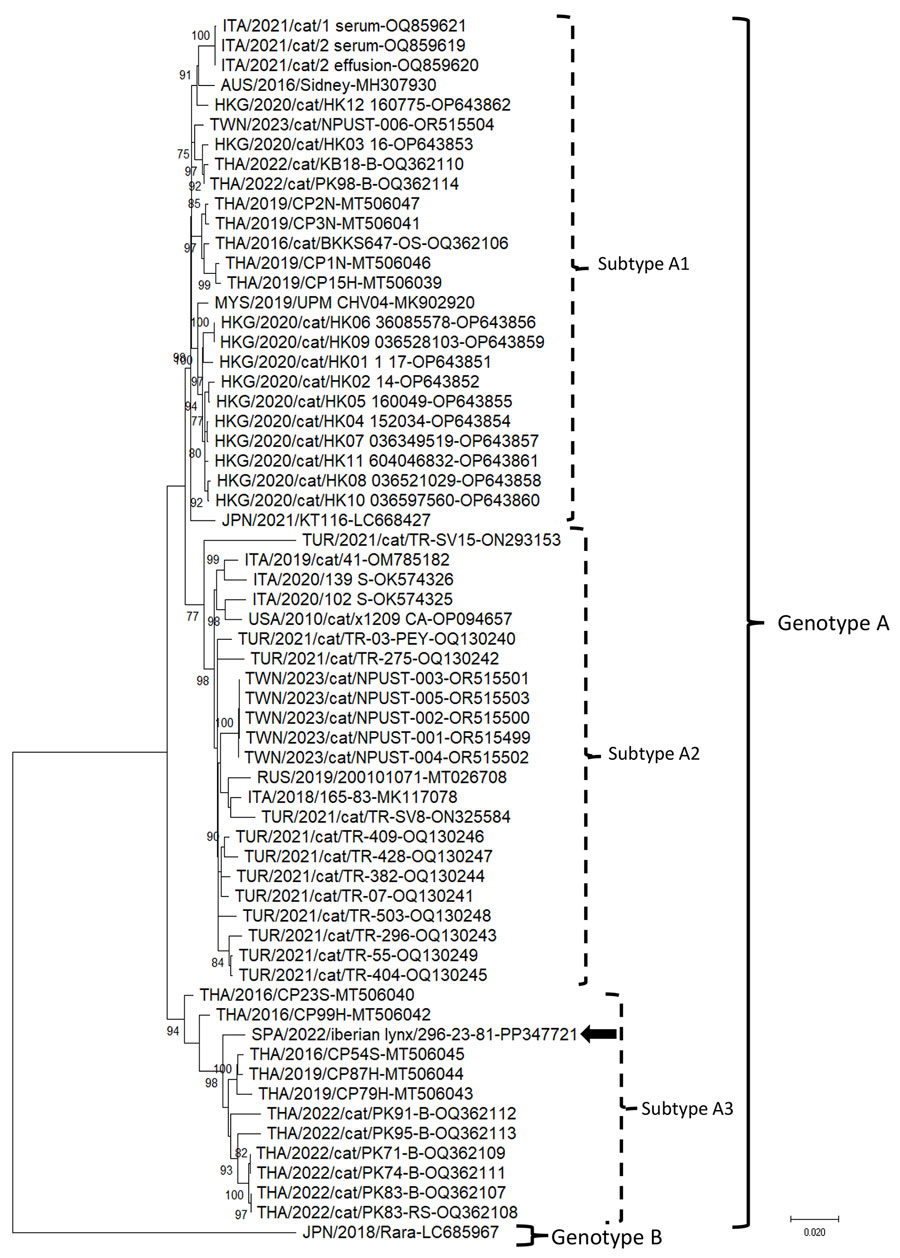
Figure. Neighbor-joining phylogenetic tree based on the complete genome of domestic cat hepadnavirus (DCH) from a study of DCH infection in Iberian lynxes. We elaborated the tree by using the alignment of the full-length nucleotide sequence of the Iberian lynx DCH SPA/2022/Iberian lynx/296-23-81 strain (black arrow; GenBank accession no. PP347721) generated in this study and the cognate sequences of DCH strains retrieved from GenBank (accession numbers shown). We constructed the tree by using the maximum-likelihood method, the Kimura 2-parameter model, a discrete gamma distribution and a proportion of invariant sites, and bootstrapping up to 1,000 replicates. Bootstrap values >70% are shown. We used the Japanese Rara strain (GenBank accession no. LC685967), belonging to DCH genotype B, as an outgroup. Scale bar indicates number of nucleotide substitutions per site.
Main Article
Page created: December 02, 2024
Page updated: December 22, 2024
Page reviewed: December 22, 2024
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.