Disclaimer: Early release articles are not considered as final versions. Any changes will be reflected in the online version in the month the article is officially released.
Volume 31, Number 6—June 2025
Research
Genesis and Spread of Novel Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus Genotype EA-2023-DG Reassortant, Western Europe
Abstract
In Europe, highly pathogenic avian influenza (HPAI) virus circulates in avian wildlife, undergoing frequent reassortment, sporadic introductions in domestic birds, and spillover to mammals. An H5N1 clade 2.3.4.4b reassortant, EA-2023-DG, affecting wild and domestic birds was detected in western Europe in November 2023. Six of its RNA segments came from the EA-2021-AB genotype, but the polymerase basic 2 and polymerase acidic segments originated from low pathogenicity avian influenza viruses. Discrete phylogeographic analyses of concatenated genomes and single polymerase basic 2 and polymerase acidic segments suggested reassortment in summer 2023 near the southwestern Baltic Sea. Subsequent continuous phylogeographic analysis of all concatenated EA-2023-DG genomes highlighted circulation in northwestern Europe until June 2024 and long-distance dispersal toward France, Norway, England, Slovakia, Switzerland, and Austria. Those results illustrate the value of phylodynamic approaches to investigate emergence of novel avian influenza virus variants, trace their subsequent dispersal history, and provide vital clues for informing outbreak prevention and intervention policies.
Since 2016, Europe has experienced periodic introductions of clade 2.3.4.4b highly pathogenic avian influenza (HPAI) subtype H5 viruses via wild migratory bird movements and sporadic spillover events to poultry, which have resulted in several outbreaks affecting wildlife and poultry (1). In 2021, the typically seasonal epidemiologic cycle changed, and HPAI viruses became enzootic in Europe (2). The hemagglutinin (HA) gene of HPAI clade 2.3.4.4 viruses (3) showed rapid wild bird–mediated expansion, including global spread (4), and rapid evolution in all affected geographic areas (5–7). After 2021, increased HPAI incidence in Europe resulted in greater genetic diversity and in the evolution of new genotypes by frequent reassortment events. By 2022, the continued circulation of HPAI viruses led to numerous poultry outbreaks, affected an increasing number of wild bird species, and led to frequent reassortment, making assessment of the epidemiologic situation difficult (2). Sporadic infection of mammals is observed, including the unexpected spread to cattle in North America and cases of human infection (8,9), and is sometimes associated with adaptive mutations for viral replication in mammalian cells. Those observations stress the zoonotic risk associated with this particular HPAI virus (HPAIV) clade and the need for surveillance efforts, including whole-genome sequencing.
By the end of 2023, continued circulation and frequent reassortment events resulted in the co-circulation of 11 H5Nx HPAIV genotypes in Europe, 7 of which emerged during the fall of 2023 (10,11). One of those novel reassortants was assigned genotype EA-2023-DG, according to the standardized avian influenza virus (AIV) genotyping nomenclature in Europe (2). EA-2023-DG was first reported in Germany in November 2023 and shared most of its genomic segments with the contemporary dominating genotype EA-2021-AB but also gained polymerase basic protein (PB) 2 and polymerase acidic protein (PA) segments from low pathogenicity avian influenza viruses circulating in western Europe (10). Using all available EA-2023-DG genotype complete genome sequences, we conducted recombination and discrete as well as continuous phylogeographic analyses to investigate the emergence and subsequent dispersal dynamic of this genotype.
Data Selection and Genotyping
The European Union Reference Laboratory, national reference laboratories, and some other partners perform sequencing analyses of the complete genome of HPAI H5Nx viruses detected through ongoing surveillance programs. All sequences are deposited in the GISAID EpiFlu database (http://www.gisaid.org). Partners perform genotyping on the basis of the phylogenetic tree topology, using previously described methods (2). During November 1, 2023–June 25, 2024, genotyping identified a total of 54 virus genomes as belonging to genotype EA-2023-DG. We extracted publicly available complete genomes comprising 8 segments from GISAID EpiFlu on January 15, 2025, resulting in a dataset consisting of all 54 identified EA-2023-DG genomes associated with exact temporal and spatial sampling metadata (Appendix 1 Table 1). The earliest publicly available EA-2023-DG genome, A/Gallus_gallus/Belgium/11307_0002/2023 (GISAID EpiFlu accession no. EPI_ISL_18607170), was sequenced from a chicken sample collected in Belgium on November 30, 2023. We used that strain as the reference strain for genotype EA-2023-DG in this study.
Investigation of Geographic Origin of Reassortant EA-2023-DG Genotype
To retrieve a selection of the most homologous sequences to each of the EA-2023-DG viral genome segments, we conducted a search in GISAID EpiFlu on May 27, 2024. For each of the genome segments of reference strain A/Gallus_gallus/Belgium/11307_0002/2023, we identified the 500 sequences with highest nucleotide homology and downloaded the sequences of all 8 gene segments of those genomes along with any available metadata (Appendix 2). The accompanying metadata included sampling location and precise collection date.
We used MAFFT 7.453 (12) to align sequences. Because genotyping identified EA-2023-DG as a reassortant containing the PB1, HA, nucleoprotein (NP), neuraminidase (NA), matrix protein (MP), and nonstructural (NS) protein segments from EA-2021-AB–like viruses, as well as PB2 and PA segments of other circulating AIVs, we assembled 3 distinct datasets. One dataset was an alignment concatenating the 6 nonrecombinant segments, PB1, HA, NP, NA, MP, and NS, of all 54 EA-2023-DG samples and the 500 most homologous H5N1 virus samples found for those 6 segments. We removed duplicates from the 6 segment searches by using a custom Python script (Python Software Foundation, https://www.python.org), resulting in a total dataset of 958 samples. The second and third datasets, 1 for PA and 1 for PB2, used a distinct PA or PB2 alignment from all 54 EA-2023-DG samples and the 500 most homologous samples for the corresponding segment.
Because the initial concatenated alignment was computationally too large to conduct Bayesian phylogeographic inference, we conducted a preliminary maximum-likelihood phylogenetic inference in IQTREE 1.6.12 (13), using default settings and the best-fitting substitution model identified by ModelFinder (14). We used that preliminary phylogenetic inference to restrict the alignment to a large monophyletic clade of 225 samples containing the 54 EA-2023-DG samples. Thus, that phylogeny-guided downsampling reflects the evolutionary history of the larger set of 958 samples.
To assess the presence of a recombination signal within the resulting PB1-HA-NP-NA-MP-NS concatenated alignment of those 6 segments, we performed the Φ test (15) implemented in the SplitsTree 4.14.8 program (16), which confirmed the occurrence of past recombination events (p<0.001). We then used the RDP4 program (17) to identify recombinant samples: we identified and discarded 41 recombinants within the set of background samples, which resulted in a PB1-HA-NP-NA-MP-NS concatenated alignment of 184 samples. We performed a new Φ test on that final alignment and confirmed the absence of a remaining recombination signal.
We conducted a discrete phylogeographic analysis based on each of the 3 alignments (PA, PB2, and recombinant-free PB1-HA-NP-NA-MP-NS) to reconstruct the transition history of viral lineages among countries and investigate the country from which EA-2023-DG genotype emerged. For those analyses, the selection of countries as discrete locations was imposed by the lack of higher sampling precision for most publicly available background sequences retrieved from GISAID EpiFlu. We performed the discrete phylogeographic analyses by using the discrete diffusion model (18) implemented in the BEAST 1.10 software package (19), specifying a GTR+Γ (general time-reversible with a gamma-distributed rate heterogeneity) nucleotide substitution model (20), a relaxed molecular clock with an underlying log-normal distribution to model branch-specific evolutionary rates (21), and a skygrid coalescent model for the tree prior (22). We ran the PA analysis for 300 Markov chain Monte Carlo (MCMC) iterations, the PB2 analysis for 500 MCMC iterations, and the concatenated analyses for 210 million MCMC iterations and sampled posterior trees every 100,000 iterations. We assessed the MCMC convergence and mixing by using the Tracer 1.7 program (23), checking that all estimated parameters were associated with an effective sample size value >200. After discarding the initial 10% of sampled posterior trees as burn-in, we retrieved and annotated the maximum clade credibility (MCC) tree by using TreeAnnotator 1.10 (19) and eventually plotted the tree in R (The R Project for Statistical Computing, https://www.r-project.org) by using a custom script (https://github.com/sdellicour/ea-2023-dg_emergence).
Reconstruction of Reassortant EA-2023-DG Dissemination History
To reconstruct the spread of the EA-2023-DG genotype after the reassortment event at its genesis, we performed a continuous phylogeographic reconstruction on the basis of the concatenated alignment of all 8 genomic segments of the 54 EA-2023-DG genomes available on January 15, 2025. We then aligned sequences again using MAFFT 7.453 (12) and performed the Φ test (15) in SplitsTree (16) to confirm the absence of a recombination signal within the concatenated alignment made of EA-2023-DG genomic sequences. The availability of precise sampling coordinates for all considered samples in this alignment made a spatially explicit phylogeographic reconstruction possible. We performed that continuous phylogeographic reconstruction by using the relaxed random walk model (24,25) in BEAST version 1.10 (19). As with the discrete approach, the continuous phylogeographic approach involves a joint inference of both the phylogenetic tree representing the evolutionary relationships between sampled sequences, and the locations, in this case the geographic coordinates (latitude and longitude) of unsampled common ancestors (26). Specifically, we used a gamma distribution to model the among-branch heterogeneity in diffusion velocity, and modeled branch-specific evolutionary rates according to a relaxed molecular clock with an underlying log-normal distribution and the nucleotide substitution process according to a GTR+Γ parameterization. As for the tree prior, we specified a flexible skygrid model (22). We ran the MCMC for 250 million iterations, sampling posterior trees every 100,000 iterations, and eventually discarded the first 25 million sampled trees as burn-in. We used Tracer 1.7.2 to assess the MCMC convergence and mixing properties and ensure that estimated parameters were all associated with an effective sample size >200. We used TreeAnnotator version 1.10 to identify and annotate the MCC tree and R functions in the SERAPHIM package (27,28) to extract the spatiotemporal information embedded within the 1,000 trees sampled from the post–burn-in posterior distribution and to estimate the weighted diffusion coefficient (29) associated with the spread of the EA-2023-DG genotype.
Epidemiologic Findings
Figure 1

Figure 1. Discrete phylogeographic analysis from study of genesis and spread of novel highly pathogenic avian influenza A(H5N1) clade 2.3.4.4b virus genotype EA-2023-DG reassortant, western Europe. The maximum clade credibility (MCC) tree...
Our analysis showed that the EA-2023-DG genotype was initially detected in a sample from a swan found dead on a Baltic Sea island in Finland on November 1, 2023 (GISAID accession no. EPI_ISL_19409779) (Figure 1; Appendix 1 Table 1). Then, on November 17, 2023, the EA-2023-DG genotype emerged in a hobby farm near the North Sea coast in northern Germany that had a mixed population of 52 chickens, 3 turkeys, and 11 ducks (Appendix 1 Table 1). EA-2023-DG subsequently spread to a total of 11 countries, and 54 cases were detected (Appendix 1 Table 2). The most affected countries included Germany, Sweden, and Poland. Germany was most affected, with cases in 13 poultry farms, 8 wild birds, 1 captive bird, and 1 wild mammal (red fox). Sweden experienced cases on 1 poultry farm and in 11 wild birds, and Poland had 5 poultry and 3 wild bird cases. Affected poultry were among the Phasianidae family (n = 21), which includes chickens (Gallus gallus) and turkeys (Meleagris gallopavo), and the Anatidae family (n = 1), which includes geese and ducks. Domestic poultry were only infected in Germany, Poland, Sweden, and Belgium. Captive animals included 1 captive barn owl (Tytonidae family, Tyto alba) and 1 black necked swan (Anatidae family, Cygnus melancoryphus) in a zoo. Wild birds, all found dead during passive surveillance efforts, included birds from the Anatidae (n = 24), Accipitridae (n = 3), Falconidae (n = 1), Strigidae (n = 1), Gruidae (n = 1), and Ardeidae (n = 1) families (Appendix 1 Table 1). A single mammal sample from a red fox (Vulpes vulpes) from the Hamburg district of Germany was EA-2023-DG positive. The most recent (June 25, 2024) occurrence was a wild goose sample in Germany.
Genesis of AIV Genotype EA-2023-DG in Western Europe
The discrete phylogeographic analyses conducted on the recombinant-free PB1-HA-NP-NA-MP-NS concatenated alignment and the PB2 and PA alignments confirmed the monophyletic nature of the EA-2023-DG clade (Figure 1; Appendix 1 Figures 1, 2). The analysis based on the PB1-HA-NP-NA-MP-NS concatenated alignment inferred the origin of the EA-2023-DG clade at the end of July 2023 (July 22, 2023; 95% highest posterior density [HPD] interval June 10–August 27, 2023) in Sweden with a posterior probability >0.99 (Figure 1). The analyses of the PB2 and PA segments, inferred its origin in Germany (posterior probability = 0.99); the PB2 analysis inferred its origin on January 6, 2023 (95% HPD September 17, 2022–April 20, 2023), and the PA analysis inferred its origin on June 30, 2023 (95% HPD April 14–September 24, 2023) (Appendix 1 Figures 1, 2).
Figure 2
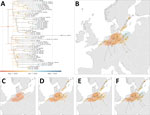
Figure 2. Continuous phylogeographic analysis from study of genesis and spread of novel highly pathogenic avian influenza A(H5N1) clade 2.3.4.4b virus genotype EA-2023-DG reassortant, western Europe. A) Time-scaled maximum clade credibility treeretrieved...
Outcomes of discrete phylogeographic inference are known to be notably affected by sampling bias (30). Thus, the discrepancy between the country of origin inferred from the discrete phylogeographic analyses conducted for the recombinant-free PB1-HA-NP-NA-MP-NS concatenated alignment on the one hand and on the PB2 and PA alignments on the other could arise from the heterogeneous sampling of closely related sequences in the different countries within the study area. However, those results overall indicated that EA-2023-DG emerged during the spring and summer of 2023 in the southwestern Baltic Sea region. The continuous phylogeographic analysis further confirms the emergence of the reassortant genotype in this region and time frame (Figure 2).
Spread History and Dynamics of EA-2023-DG
Our continuous phylogeographic analysis using concatenated EA-2023-DG sequences for all 8 segments confirmed that the ancestral node of this clade traces back to the southwestern Baltic Sea shores of Sweden, Germany, and Denmark (Figure 2, panel B). Although the most ancestral nodes of the MCC tree are inferred in northern Germany (Figure 2, panel C), the uncertainty associated with the Bayesian phylogeographic inference (shaded 80% HDP polygons) highlights a potential area of origin that is relatively large and corresponds to the larger southwestern Baltic Sea area, which aligns with the results of the discrete phylogeographic reconstructions. Our continuous phylogeographical analysis dates the most ancestral node of the EA-2023-DG clade as August 2, 2023 (95% HPD July 16–September 7, 2023), also aligning with the discrete phylogenetic analyses (Figure 2, panel A). The continuous phylogeographic analysis indicated that after its emergence, lineages of the EA-2023-DG genotype further spread in Germany and Poland, toward the Netherlands, and to southern Sweden and southern Finland before November 2023. Its lineages then reached Belgium, England, Slovakia, and Switzerland during November and December 2023. By June 2024, EA-2023-DG spread as far north as the island of Åland in south Finland; as far south as Lyon, France; as far east as Slovakia; and as far west as England (Figure 2, panels B–F). Overall, the virus predominantly circulated in Germany, Sweden, and Poland, with only occasional detections in other countries in Europe.
The ongoing panzootic caused predominantly by clade 2.3.4.4b HPAIVs is notorious for its diversifying evolution, including frequent reassortment events that result in an ever-changing range of circulating genotypes (2,6). Reassortment events represent crucial shifts in virus evolution that can affect host range, pathogenicity, and other epidemiologically relevant aspects of the virus phenotype; thus, understanding the dynamics behind the emergence and spread of such novel reassortants is critical. Combining complete avian influenza genomes and exact spatial and temporal sampling data enables detailed reconstruction of virus dispersal during an outbreak (31) and identification of reassortment events (2).
In this study, we analyzed all available full-genome sequences of novel reassortant HPAIV H5N1 genotype EA-2023-DG, which emerged in 2023 in western Europe (10), to reconstruct its genesis and dispersal dynamics. We traced its origin to the southwestern Baltic Sea area in the spring and summer of 2023. More precisely, most of the genome (i.e., PB1, HA, NP, NA, MP, and NS segments) originated from the dominant EA-2021-AB genotype, and the most recent common ancestor of those EA-2023-DG genomic segments likely emerged in or close to Sweden during summer 2023. As for the PA and PB2 segments, we inferred their origin in Germany, meaning that they could have originated from low pathogenicity avian influenza viruses circulating in Germany during winter and spring 2023, as suggested by others merely on the basis of sequence similarity (10). Overall, our results point toward a local reassortment event that occurred in the southwestern Baltic Sea area, which is in line with the first occurrence of the genotype in southern Finland.
Of note, our phylogeographic reconstructions of the genesis of EA-2023-DG agree with the AIV introduction risk prediction on the basis of wild bird migration data and AIV occurrences by the EFSA Bird Flu Radar Tool (32). Those predictions indicated high introduction risk near the southwestern Baltic Sea along the shores of Germany, Denmark, and Sweden for the week fitting the time to most recent common ancestor (tMRCA) of the EA-2023-DG clade (August 7–13, 2023). In addition, the population density and ringing recapture data of species from the Anatidae family associated with the most distant translocations of EA-2023-DG and captured in Germany and Denmark around the week of the tMRCA according to the Migration Mapping Tool, Bird Flu Radar Tool (32), do not contradict the viral lineage movements we reconstructed here. Those observations confirm the continued involvement of an increasing spectrum of wild bird species in the epidemiology of HPAIV.
The spectrum of bird families affected by EA-2023-DG, including Phasianidae, Anatidae, Accipitridae, Falconidae, Strigidae, Tytonidae, Gruidae, and Ardeidae, aligns largely to the host spectrum of genotype EA-2021-AB that donated most of the genome (2). A single dead fox was found infected in the core area of EA-2023-DG circulation in northern Germany, confirming the role of wild carnivores as dead-end hosts of HPAIV clade 2.3.4.4b (5). Because of the reassortment event, we could not include ancestral DG precursor genomes in the detailed whole-genome–based phylogeographic reconstruction, resulting in a substantial spatial uncertainty covering the western Baltic Sea shores and North Sea coast of Germany. Another factor that might have contributed to the temporal and spatial uncertainty in our predictions is the lack of standardized surveillance approaches between countries, especially for wildlife surveillance. Our spatially explicit phylogeographic reconstruction highlights continued circulation with a focus in Germany, Poland, and Sweden and sporadic occurrences as far north as central Sweden, as far south as central France, as far west as England, and as far east as Slovakia.
During its period of circulation, EA-2023-DG became the second most frequent (54 cases) genotype in the countries it affected, but EA-2021-AB remained the dominant genotype with 84 reported cases. Other prevalent genotypes were EA-2023-DB (32 cases), EA-2024-DI (26 cases), EA-2022-BB (16 cases), EA-2021-I (14 cases), and EA-2023-DA (13 cases) (33,34). Ten additional genotypes circulated at lower frequency (<10 cases), reflecting the known diversification potential of H5N1 clade 2.3.4.4b viruses (2).
In vivo experiments following up on the emergence of HPAIV H5N1 in cattle in the United States (35), and its subsequent spillover to other mammals, including cats (35) and exposed humans (36), used an EA-2023-DG genotype virus as a model of contemporary circulating viruses in Europe. Those studies indicated that these viruses efficiently replicate in bovine mammary tissue and can produce adaptive mutations (PB2 E627K) during replication (37). Those findings underscore the value of phenotypic characterization of currently circulating H5Nx clade 2.3.4.4b viruses, including the newly emerged EA-2023-DG genotype, because the zoonotic potential of the viruses can evolve, driven and shaped by epidemiologic events that could increase the likelihood of spillover to mammals and subsequent adaptation. In response to HPAIV reassortment promiscuity resulting in fast evolution and diversification (2,6), efficient livestock and wildlife surveillance programs including a viral genomic characterization are essential.
In conclusion, although gaps in surveillance data will always exist, we demonstrated that viral genomic data collected from surveillance programs combined with precise spatial and temporal metadata can enable a comprehensive investigation of the genesis of novel AIV reassortants and of their spread dynamics. In addition to viral genetic characterization, such as adaptive mutations and genotyping, these parameters provide vital clues for informing outbreak prevention and intervention policies.
Dr. Van Borm is a senior scientist at the Avian Virology and Immunology Unit of Sciensano, Brussels, Belgium. His research interests include molecular diagnostics, characterization, and evolution of livestock viruses including avian influenza and Newcastle disease virus.
Author contributions: S.V.B.: conceptualization, investigation, formal analysis, methodology, supervision, data curation, writing original draft, writing review, and editing. S.D.: conceptualization, investigation, formal analysis, methodology, visualization, supervision, writing original draft, writing review, and editing. A.K.A., C.B., A.C.B., C.A.B., F.B., Z.D., M.E., A.F., E.G., B.Gj., B.Gr., P.H., A.K., B.L., B.C.M., I.M., A.N., A.P., D.P., S.M.R., S.R., M.Ste., M.Stä., E.S., N.T., M.W., B.Z., S.Z.: investigation, resources, data curation, methodology, writing review, and editing.
Acknowledgments
We thank K. De Geest for data curation, and all staff and organizations involved in surveillance activities in the affected countries, including L. Palumbo and the French Biodiversity Agency and I. Mertens and the Federal Agency for the Safety of the Food Chain, Brussels. We also gratefully acknowledge the authors of originating and submitting laboratories for sequences from GISAID’s EpiFlu Database used in this research (Appendix 2).
Influenza A virus genomic sequences used in this study are available in the GISAID EpiFlu database (accession numbers listed in Appendix 1 Table 1; Appendix 2). R scripts related to the preparation and visualization of discrete and continuous phylogeographic analyses are available along with the associated input files at https://github.com/sdellicour/ea-2023-dg_emergence.
Support was provided by the European Commission within the framework of the activities foreseen by the European Union Reference Laboratory for Avian Influenza and Newcastle Disease under grant agreement no. SI2.870510, as well as by the Belgian Federal Public Service Health, Food Chain Safety and Environment under grant agreement no. RT 23/01 FLUCART 1. Support by the European Union under the Horizon Europe programme grant agreement KAPPA-FLU no. 101084171 is also acknowledged. A.P. and A.K.A. acknowledge support by the German Federal Ministry of Education and Research within the project 'PREPMEDVET' grant no. 13N15449. A.C.B., S.R., and B.M. were partially funded by the UK Department for the Environment, Food and Rural Affairs (Defra) and the devolved Scottish and Welsh governments under grant nos. SV3032, SV3400, and SE2227. S.D. acknowledges support from the Fonds National de la Recherche Scientifique (F.R.S.-FNRS, Belgium; grant no. F.4515.22), from the Research Foundation—Flanders (Fonds voor Wetenschappelijk Onderzoek — Vlaanderen, FWO, Belgium; grant no. G098321N), and from the European Union Horizon 2020 projects MOOD (grant agreement no. 874850) and LEAPS (grant agreement no. 101094685). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or REA. Neither the European Union nor the granting authority can be held responsible for them.
References
- Verhagen JH, Fouchier RAM, Lewis N. Highly pathogenic avian influenza viruses at the wild-domestic bird interface in Europe: future directions for research and surveillance. Viruses. 2021;13:212. DOIPubMedGoogle Scholar
- Fusaro A, Zecchin B, Giussani E, Palumbo E, Agüero-García M, Bachofen C, et al. High pathogenic avian influenza A(H5) viruses of clade 2.3.4.4b in Europe-Why trends of virus evolution are more difficult to predict. Virus Evol. 2024;10:
veae027 . DOIPubMedGoogle Scholar - Smith GJD, Donis RO; World Health Organization/World Organisation for Animal Health/Food and Agriculture Organization (WHO/OIE/FAO) H5 Evolution Working Group. Nomenclature updates resulting from the evolution of avian influenza A(H5) virus clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013-2014. Influenza Other Respir Viruses. 2015;9:271–6. DOIPubMedGoogle Scholar
- Antigua KJC, Choi W-S, Baek YH, Song M-S. The emergence and decennary distribution of clade 2.3.4.4 HPAI H5Nx. Microorganisms. 2019;7:156. DOIPubMedGoogle Scholar
- Graziosi G, Lupini C, Catelli E, Carnaccini S. Highly pathogenic avian influenza (HPAI) H5 clade 2.3.4.4b virus infection in birds and mammals. Animals (Basel). 2024;14:1372. DOIPubMedGoogle Scholar
- Kandeil A, Patton C, Jones JC, Jeevan T, Harrington WN, Trifkovic S, et al. Rapid evolution of A(H5N1) influenza viruses after intercontinental spread to North America. Nat Commun. 2023;14:3082. DOIPubMedGoogle Scholar
- Lee D-H, Criado MF, Swayne DE. Pathobiological origins and evolutionary history of highly pathogenic avian influenza viruses. Cold Spring Harb Perspect Med. 2021;11:
a038679 . DOIPubMedGoogle Scholar - Peacock TP, Moncla L, Dudas G, VanInsberghe D, Sukhova K, Lloyd-Smith JO, et al. The global H5N1 influenza panzootic in mammals. Nature. 2025;637:304–13. DOIPubMedGoogle Scholar
- Webby RJ, Uyeki TM. An update on highly pathogenic avian influenza A(H5N1) virus, clade 2.3.4.4b. J Infect Dis. 2024;230:533–42. DOIPubMedGoogle Scholar
- Ahrens AK, Pohlmann A, Grund C, Harder T, Beer M. Novel genotypes of highly pathogenic avian influenza H5N1 clade 2.3.4.4b viruses, Germany, November 2023. Emerg Infect Dis. 2024;30:1737–9. DOIPubMedGoogle Scholar
- Adlhoch C, Fusaro A, Gonzales JL, Kuiken T, Mirinavičiūtė G, Niqueux É, et al.; European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza. Avian influenza overview September-December 2023. EFSA J. 2023;21:
e8539 .PubMedGoogle Scholar - Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. DOIPubMedGoogle Scholar
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. DOIPubMedGoogle Scholar
- Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–81. DOIPubMedGoogle Scholar
- Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. DOIPubMedGoogle Scholar
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1:
vev003 . DOIPubMedGoogle Scholar - Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLOS Comput Biol. 2009;5:
e1000520 . DOIPubMedGoogle Scholar - Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. 1. Virus Evol. 2018;4:
vey016 . DOIPubMedGoogle Scholar - Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994;11:725–36.PubMedGoogle Scholar
- Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:
e88 . DOIPubMedGoogle Scholar - Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol Biol Evol. 2013;30:713–24. DOIPubMedGoogle Scholar
- Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67:901–4. DOIPubMedGoogle Scholar
- Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol. 2010;27:1877–85. DOIPubMedGoogle Scholar
- Pybus OG, Suchard MA, Lemey P, Bernardin FJ, Rambaut A, Crawford FW, et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc Natl Acad Sci U S A. 2012;109:15066–71. DOIPubMedGoogle Scholar
- Dellicour S, Gill MS, Faria NR, Rambaut A, Pybus OG, Suchard MA, et al. Relax, keep walking—a practical guide to continuous phylogeographic inference with BEAST. Mol Biol Evol. 2021;38:3486–93. DOIPubMedGoogle Scholar
- Dellicour S, Rose R, Pybus OG. Explaining the geographic spread of emerging epidemics: a framework for comparing viral phylogenies and environmental landscape data. BMC Bioinformatics. 2016;17:82. DOIPubMedGoogle Scholar
- Dellicour S, Rose R, Faria NR, Lemey P, Pybus OG. SERAPHIM: studying environmental rasters and phylogenetically informed movements. Bioinformatics. 2016;32:3204–6. DOIPubMedGoogle Scholar
- Trovão NS, Suchard MA, Baele G, Gilbert M, Lemey P. Bayesian inference reveals host-specific contributions to the epidemic expansion of influenza A H5N1. Mol Biol Evol. 2015;32:3264–75. DOIPubMedGoogle Scholar
- Layan M, Dacheux L, Lemey P, Brunker K, Ma L, Troupin C, et al. Uncovering the endemic circulation of rabies in Cambodia. Mol Ecol. 2023;32:5140–55. DOIPubMedGoogle Scholar
- Van Borm S, Boseret G, Dellicour S, Steensels M, Roupie V, Vandenbussche F, et al. Combined phylogeographic analyses and epidemiologic contact tracing to characterize atypically pathogenic avian influenza (H3N1) epidemic, Belgium, 2019. Emerg Infect Dis. 2023;29:351–9. DOIPubMedGoogle Scholar
- Gargallo G, Davies JG, Faverjon C, Kampichler C, Baillie SR, Cameron A, et al. Development of a prototype early warning system for avian influenza in the EU based on risk-mapping. EFSA Support Publ. 2022;19:7762E. DOIGoogle Scholar
- Fusaro A, Gonzales JL, Kuiken T, Mirinavičiūtė G, Niqueux É, Ståhl K, et al.; European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza. Avian influenza overview December 2023-March 2024. EFSA J. 2024;22:
e8754 .PubMedGoogle Scholar - Alexakis L, Fusaro A, Kuiken T, Mirinavičiūtė G, Ståhl K, Staubach C, et al.; European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza. Avian influenza overview March-June 2024. EFSA J. 2024;22:
e8930 .PubMedGoogle Scholar - Burrough ER, Magstadt DR, Petersen B, Timmermans SJ, Gauger PC, Zhang J, et al. Highly pathogenic avian influenza A(H5N1) clade 2.3.4.4b virus infection in domestic dairy cattle and cats, United States, 2024. Emerg Infect Dis. 2024;30:1335–43. DOIPubMedGoogle Scholar
- Garg S, Reed C, Davis CT, Uyeki TM, Behravesh CB, Kniss K, et al. Outbreak of highly pathogenic avian influenza A(H5N1) viruses in U.S. dairy cattle and detection of two human cases—United States, 2024. MMWR Morb Mortal Wkly Rep. 2024;73:501–5. DOIPubMedGoogle Scholar
- Halwe NJ, Cool K, Breithaupt A, Schön J, Trujillo JD, Nooruzzaman M, et al. H5N1 clade 2.3.4.4b dynamics in experimentally infected calves and cows. Nature. 2025;637:903–12. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: May 05, 2025
Table of Contents – Volume 31, Number 6—June 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Steven Van Borm, Sciensano, Avian Virology and Immunology, Groeselenberg 99, B1180 Ukkel, Brussels B1180, Belgium
Top