Volume 31, Number 7—July 2025
Research
Epidemiologic and Genomic Investigation of Sexually Transmitted Shigella sonnei, England
Cite This Article
Citation for Media
Abstract
Shigellosis is a bacterial infection that causes enteric illness and can be sexually transmitted, particularly among gay, bisexual, and other men who have sex with men. Multiple extensively drug-resistant Shigella strains have been detected through genomic surveillance and are associated with plasmids carrying the gene variant blaCTX-M-27 in the United Kingdom. We report an increase in possible sexually transmitted cases of Shigella bacteria carrying the blaCTX-M-15 gene variant, which was previously associated with travel. In 2023, there were 117 cases belonging to the 10 single-nucleotide polymorphism linkage cluster t10.1814. Although this cluster has been documented in England since August 2019, genetic analyses revealed that the blaCTX-M-15 gene variant entered the lineage on a novel resistance plasmid coinciding with the first outbreak case. Our analysis highlights the shifting antimicrobial resistance landscape of sexually transmitted Shigella bacteria. Parallel emergence of resistance determinants against third-generation cephalosporins in sexual transmission networks suggests high levels of antimicrobial selection pressure.
Shigellosis is a gastrointestinal infection caused by 1 of 4 bacterial species, Shigella sonnei, S. flexneri, S. boydii, or S. dysenteriae. Common symptoms include bloody diarrhea, abdominal pain, cramps, fever, nausea, and vomiting (1). Shigella spp. are anthroponotic and transmitted by fecal–oral contact (1), from hands or objects that were in contact with human feces, including through sexual contact (2). Infection can also occur through contaminated food and water (3,4) or travel to endemic countries (5). Community outbreaks are associated with childcare settings, schools, residential institutions, and restaurants (6–8). Persons at highest risk for infection include those attending childcare settings, travelers to endemic countries, and gay, bisexual, and other men who have sex with men (GBMSM).
The implementation of whole-genome sequencing (WGS) for public health surveillance of bacterial pathogens has enabled global monitoring of the emergence and transmission of epidemic strains of Shigella spp. and antimicrobial resistance. Antimicrobial-resistant S. sonnei were first described >60 years ago (9–11), and multidrug-resistant (MDR) strains resistant to aminoglycosides, sulphonamides, trimethoprim, or chloramphenicol are endemic in the human population on every continent (12). Resistance to fluroquinolones has recently emerged, including from regions where antimicrobial use is unregulated (13–15). The increasing incidence of MDR and extensively drug-resistant (XDR) shigellosis in high-prevalence regions where surveillance is limited can be monitored by sequencing strains of S. flexneri and S. sonnei isolated from returning travelers and analyzing the genome derived antimicrobial resistance profiles.
Since 2010, surveillance systems maintained by the United Kingdom Health Security Agency (UKHSA) have identified a series of epidemics of MDR S. flexneri serotypes 3a, 2a, and 1b and S. sonnei among GBMSM; the strains are circulating nationally and internationally (12,16–18). Previous studies have demonstrated the acquisition of a plasmid encoding resistance to macrolides corresponded with the emergence of epidemics of S. flexneri 3a and 2a and S. sonnei during 2010–2015 (17,19). The subsequent global increase in notification of S. sonnei among GBMSM was enabled by strains belonging to global lineage 3.6.1.1.2 (clonal complex [CC] 152), exhibiting resistance to both macrolides and fluroquinolones (16). During the COVID-19 pandemic, a rapid decrease in notifications of S. sonnei was observed in the United Kingdom. However, after the relaxation of social distancing and travel restrictions, notifications quickly returned to prepandemic levels (20). We observed an increase in XDR S. sonnei with the blaCTX-M-27 gene variant conferring resistance to third-generation cephalosporins (21). Localized and short-lived outbreaks of XDR S. sonnei and S. flexneri containing the blaCTX-M-27 gene variant, primarily circulating within GBMSM sexual networks, were described previously (18,22). In contrast, an epidemic of sexually transmitted XDR S. sonnei was recorded in September 2021 (designated t10.377 by using the UKHSA single-linkage hierarchical clustering methodology, contained within global lineage 3.6.1.1.2 and CC152), continued into 2022 and was reported internationally (21).
After the publication of a study from France reporting an increase in the proportion of Shigella spp. isolates simultaneously resistant to ciprofloxacin, third-generation cephalosporins, and azithromycin (23), we reviewed genome-derived antimicrobial resistance profiles of the S. sonnei in the UKHSA archive isolated during 2016–2023. We identified an increasing trend of XDR strains of S. sonnei and found XDR S. sonnei isolated from MSM almost exclusively had the blaCTX-M-27 gene variant, whereas XDR S. sonnei isolated from travelers returning from high-risk regions almost exclusively had the blaCTX-M-15 gene variant (24). In 2023, we detected an increase of XDR S. sonnei in England that contained the blaCTX-M-15 gene variant. The aim of this study was to use a combination of epidemiologic data with short-read and long-read genomic sequencing data for outbreak investigation to determine emergence and transmission patterns of the S. sonnei outbreak strain and acquisition of the blaCTX-M-15 gene variant.
Routine Laboratory and Epidemiologic Surveillance
Shigella spp. isolates from hospital and community cases with gastrointestinal symptoms are referred to the gastrointestinal bacterial reference unit at the UKHSA for confirmation and typing. Since September 2015, we have conducted WGS for all Shigella isolates submitted to the gastrointestinal bacterial reference unit as previously described (25) and derived the serotype and antimicrobial resistance profile in silico from the genome. S. sonnei isolates submitted to the gastrointestinal bacterial reference unit during January 2016–December 2023 were included in this study. Because of the lack of sexual orientation information available in this dataset, we used a proxy indicator of cases that might be attributed to sexual transmission among GBMSM, defined as cases among male adults (>16 years) without a history of travel or where travel history was unknown (presumptive men who have sex with men [MSM]) (26).
We analyzed the sequencing data for genomic markers of resistance to azithromycin (defined as the presence of ermB or mphA), ciprofloxacin (defined as the presence of mutations in gyrA, parC, or qnr), and third-generation cephalosporins (defined by the presence of blaCTX-M genes). We defined XDR isolates as those containing genomic markers of resistance to azithromycin, ciprofloxacin, and third-generation cephalosporins.
We conducted single-nucleotide polymorphism (SNP) typing on S. sonnei isolates. We applied single-linkage hierarchical clustering at 7 descending thresholds of SNP distances (Δ250, Δ100, Δ50, Δ25, Δ10, Δ5, Δ0) as previously described (26). That clustering resulted in a discrete 7-digit code in which each number represents the cluster membership at each descending SNP distance threshold. For Shigella spp. surveillance, we designated isolates that cluster at the 10 SNP threshold t10.X. We duplicated sequencing data in line with routine genomic surveillance of Shigella spp. at UKHSA. We tested the differences in proportions by using 2-proportion Z-tests and defined p<0.05 as significant.
Phylogenetic Tree Construction
We used the WGS data from routine laboratory surveillance to create a phylogenetic tree of S. sonnei isolates with blaCTX-M gene variants. We produced 1,325 samples from a soft-core genome alignment of CC152 within nucleotide cluster t25:1, in which a given variant position belonged to <80% of strains in the alignment, by using SnapperDB v0.2.8 (27). We previously masked recombinant sequences from a whole genome alignment derived from SnapperDB v0.2.8 (27) on the same dataset by using Gubbins v3.2 (28). We used the alignment (2,142,354 bp) as the input for IQ-TREE v2.0.4 (29) to generate a phylogenetic tree. We then repeated the methodology to produce subtrees of each cluster containing genomes with blaCTX-M variants. For each phylogeny, the tree was rooted by the most closely related strain outside the cluster range in question.
Nanopore Sequencing and De Novo Assembly
We used Illumina (Illumina, https://www.illumina.com) for routine sequencing and Oxford Nanopore (Oxford Nanopore Technologies, https://nanoporetech.com) for long-read sequencing to generate complete assemblies of selected blaCTX-M variant samples to understand the genetic context for antimicrobial resistance determinants. We extracted and sequenced genomic DNA by using the MinION (Oxford Nanopore Technologies) and processed data, trimmed reads, and assembled as described previously (30).
We conducted de novo assembly by using Flye v2.9.2 (31). We corrected the assemblies by using Medaka version 1.0.3 (https://github.com/nanoporetech/medaka) with a Shigella-specific medaka-trained model, and then by using Polypolish v0.5.0 (32) with the equivalent Illumina FASTQs (Illumina) for each assembly. Because all the contigs were circular and closed, we reoriented them to start at the dnaA gene (GenBank accession no. NC_000913) from E. coli K12, by using the fix start parameter in Circlator version 1.5.5 (33).
Antimicrobial Resistance Gene Detection and Plasmid Typing
We detected the plasmid replicon for each nonchromosomal contig within the final assembly of each sample by using PlasmidFinder version 2.1 (34) with the Enterobacteriaceae database and these parameters: minimum identity = 90% and minimum coverage = 90%. We annotated the mobile genetic elements with antimicrobial resistance determinants by using the Prokaryotic Genome Annotation Pipeline build 2022-12-13 (35). We generated gene-level alignments by using Clinker version 0.0.27 (36).
Data Deposition
We submitted the FASTQ files and gene assemblies to the National Center for Biotechnology Information (BioProject no. PRJNA315192). Accession numbers have been provided (Appendix Table 1).
Ethics Statement
This study was undertaken for health protection purposes. Permission was granted to UKHSA to collect and process confidential patient data under Regulation 3 of The Health Service (Control of Patient Information) Regulations 2020 and Section 251 of the National Health Service Act 2006.
Descriptive Epidemiology
Figure 1

Figure 1. Epidemiologic and genomic investigation of sexually transmitted Shigella sonnei diagnoses in presumptive MSM classification, England, 2016–2023. Presumptive MSM category was defined as cases among male adults (>...
S. sonnei diagnoses increased during 2016–2018, then declined slightly in 2019 and declined markedly in 2020 and 2021, likely because of reduced access to healthcare services and testing, social distancing, and travel restrictions during the COVID-19 pandemic (Figure 1). In 2022 and 2023, there was a substantial increase in S. sonnei diagnosis notifications, and the 2023 notifications exceeded prepandemic levels. The trends of S. sonnei among presumptive MSM mirror those among all persons. However, the rate of increase was larger among presumptive MSM, leading to an increase in the proportion of all S. sonnei diagnoses seen among presumptive MSM, from 26% in 2016 to 46% in 2023. The increase between 2022 and 2023 was also higher among presumptive MSM (82% increase) compared with all persons (50% increase).
Figure 2
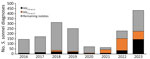
Figure 2. Sexually transmitted Shigella sonnei isolates among presumptive men who have sex with men by the presence of the blaCTX-M-15 or blaCTX-M-27gene variant...
Including the increase in S. sonnei among presumptive MSM in 2023, there was a corresponding increase in the number and proportion of S. sonnei isolates with the blaCTX-M-15 gene variant in the population. During 2016–2022, an average of 10% of S. sonnei isolates contained the blaCTX-M-15 gene variant, increasing to 33% in 2023. That increase in the proportion of S. sonnei isolates with the blaCTX-M-15 gene variant in 2023 corresponded with a decrease in S. sonnei isolates with the blaCTX-M-27 gene variant in this population group (Figure 2).
Figure 3

Figure 3. Shigella sonnei isolates with the blaCTX-M-15gene variant among presumptive men who have sex with men compared with nonpresumptive men who have sex with men from an...
Before 2023, S. sonnei isolates with the blaCTX-M-15 gene variant were identified at a much lower frequency among presumptive MSM compared with non–presumptive MSM (i.e., women, children, and men reporting recent travel). During 2016–2022, the proportion of S. sonnei with the blaCTX-M-15 gene variant among presumptive MSM remained stable at an average of 17%, increasing to 38% in 2023 (Figure 3).
Phylogenetic Analysis of S. sonnei with the blaCTX-M-15 Gene Variant
Figure 4

Figure 4. Maximum-likelihood phylogenetic tree of the Shigella sonneiclonal complex 152 within the 10 single-nucleotide polymorphism linkage cluster t10.1814 (n = 125) found during an epidemiologic and genomic investigation of...
Figure 5
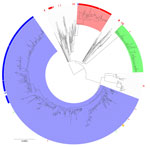
Figure 5. Maximum-likelihood phylogenetic tree of clonal complex 152 within nucleotide cluster t25:1 found during an epidemiologic and genomic investigation of sexually transmitted Shigella sonneifrom presumptive men who have sex...
Of the 262 S. sonnei isolates with the blaCTX-M-15 gene variant collected during 2016–2023 from presumptive MSM, 84 (32%) fell within a 10-SNP single linkage cluster (SCL) designated t10.1814 (full SNP address 1.1.1.1.1814) and belonging to global lineage 3.6.1.1 (37) (Figure 4). In addition, 2 other 10-SNP SCLs contained isolates with the blaCTX-M-15 gene variant were identified, t10.1148 (full SNP address 1.1.29.49.1148) and t10.2187 (full SNP address 1.1.29.49.2187). Those 2 clusters fall within the same 25-SNP SCL (t25:49), a lineage that is associated with travel to Pakistan (82% of case-patients reporting travel to Pakistan) and Tunisia (94% of case-patients reporting travel to Tunisia), but distinct from the 25-SNP SCL containing t25:1. The remaining S. sonnei isolates with the blaCTX-M-15 gene variant from presumptive MSM were phylogenetically sporadic and did not form large clusters (Figure 5). Therefore, our phylogenetic analysis focuses on the t10.1814 cluster to explore the increase in S. sonnei containing the blaCTX-M-15 gene variant among presumptive MSM (Figure 4).
Figure 6
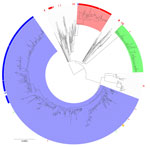
Figure 6. Diagnoses of Shigella sonnei in cluster t10.1814 by the blaCTX-M-15 gene variant status from an epidemiologic and genomic investigation, England, 2016–2023.
At the end of 2023, the t10.1814 cluster contained 124 isolates in total. The first 3 cases within the cluster were diagnosed in August and October 2019. None of those isolates contained blaCTX-M-15; however, 2 of the 3 isolates contained the blaCTX-M-27 gene variant (Figures 4,5,6). There was no reported activity within the t10.1814 cluster until March 2022, but 4 cases were reported during March–December 2022. There was a substantial increase in cases in 2023, and most isolates (94%, 117/124) in the cluster had specimen dates in 2023. Of the 2023 isolates, 92% (108/117) contained the blaCTX-M-15 gene variant (Figure 6). Of the 124 cases in the cluster, 75% (n = 93) were adult men with no or unknown travel, 16% (n = 20) were adult men with travel outside the UK (mostly to countries in Europe), and 9% (n = 11) were women or children with no or unknown travel (Table 1).
Figure 7

Figure 7. Diagnoses of Shigella sonneiin the t10.377 cluster compared with the t10.1814 cluster among presumptive men who have sex with men from an epidemiologic and genomic investigation, England, 2016–2023....
The increase in t10.1814 occurred in parallel to a decline in cases within the 1.1.1.1.377.% cluster (designated t10.377; the % indicates that t3 and t0 positions of the SNP address can take any value). The cluster was dominant since 2017, declined substantially during the first few months of the COVID-19 pandemic, but reemerged in September 2021 with third-generation cephalosporin resistance caused by the blaCTX-M-27 gene variant (21) (Figure 7). Despite the different trends in those clusters, t10.1814 and t10.377 share similarities in demographic characteristics of cases; the t10.377 cluster was also associated with presumptive MSM (Appendix Table 2).
Of the 124 isolates in the t10.1814 cluster, 112 (90%) harbored the blaCTX-M-15 gene variant, whereas only 2 isolates had the blaCTX-M-27 gene variant; no isolates expressed both the blaCTX-M-15 and blaCTX-M-27 gene variants. Resistance to ciprofloxacin and azithromycin was very high; 94% of isolates (116/124) exhibited mutations in either gyrA, parC, or the plasmid mediated qnr gene variant, and 88% of isolates (109/124) had genomic markers for azithromycin resistance (ermB or mphA). Most (86%) isolates had both the blaCTX-M-15 gene variant and markers of azithromycin resistance. Overall, 109 (88%) isolates in the t10.1814 cluster were XDR (Table 2).
Phylogenetic analysis revealed that t10.1814 fell within the wider t25:1 cluster that also includes t10.377. Although located within the same t25 SLC, the t10.1814 cluster with blaCTX-M-15 was located on a separate branch and did not evolve from the t10.377 cluster with the blaCTX-M-27 gene variant (Figure 5). The progenitor strains of t10.1814 clustered with the blaCTX-M-15 gene variant also contained the blaCTX-M-27 gene variant.
Analysis of t10.1814 IncFII Plasmids and Comparison to t10.377 IncFII Plasmids
Figure 8

Figure 8. Alignment of IncFII plasmids in samples selected for Nanopore sequencing during an epidemiologic and genomic investigation of sexually transmitted Shigella sonneifrom presumptive men who have sex with men,...
Plasmids within t10.1814 with the blaCTX-M-15 gene variant were all determined to be of the IncFII replicon type and ranged from 77.6 to 149.0 kbp in size (Figure 8). Plasmids from progenitor strains within t10.1814 with the blaCTX-M-27 gene variant were larger on average (148 kbp) than plasmids with the blaCTX-M-15 gene variant (78.4 kbp). Despite the difference in size, almost all the gene content of the ≈74 kbp plasmids were also found within the larger ≈148 kbp plasmids. Of note, when comparing t10.1814 plasmids to IncFII plasmids from t10.377, the plasmid structure is the same except for small alterations to the variable region, including the blaCTX-M-15 and blaCTX-M-27 integrons (Figure 8).
Figure 9

Figure 9. Alignment of exemplar IncFII plasmid from a Shigella sonnei strain during an epidemiologic and genomic investigation of sexually transmitted Shigella sonneifrom presumptive men who have sex...
After the analysis of the long-read sequencing data, we identified 1 isolate of S. sonnei with the blaCTX-M-15 gene variant in the same clade as t10.1814 but in a different t10 SLC (t10.2404). In that cluster, the blaCTX-M-15 gene variant was located on a 7.6-kbp cassette or an element integrated on the chromosome. The integration site appears to be a prophage remnant near a tRNA-Phe gene (Figure 9). The IncFII plasmid was lost in this sample.
Overall, except for 2020 and 2021 when notifications were affected by the COVID-19 pandemic, we observed a steady increase of S. sonnei diagnoses in England during 2016–2023. During the past decade, diagnostic methods for the detection of gastrointestinal pathogens have improved with widespread implementation of commercial PCRs. PCR is more sensitive than culture for detecting Shigella spp. (38,39), and this move toward molecular methods after culture will increase case confirmation. Furthermore, the increase might be associated with increased travel to high-risk regions outside the United Kingdom, although confirming that theory is difficult because travel history is poorly captured by the current surveillance system. We also observed a steady increase in notifications of S. sonnei among presumptive MSM. Although our observation may reflect a true increase in sexual transmission, it might also be influenced by increased implementation of PCR testing and travel. The increase in reported diagnoses might be because of the publication of briefing notes and other outbreak-related communications by UKHSA during the study period.
Numerous factors enable the emergence, transmission, and persistence of epidemic strains circulating within GBMSM sexual networks, involving pathogen characteristics, host behaviors, and environmental pressures. We have previously hypothesized that the sequential waves of shigellosis among GBMSM in the United Kingdom have been enabled by acquisition of antimicrobial resistance to an increasing number of classes of antimicrobial drugs (40). The epidemic strains of S. sonnei were initially resistant to macrolides, then to both azithromycin and ciprofloxacin, and most recently to macrolides, fluroquinolones, and third-generation cephalosporins (16,21). However, the acquisition of antimicrobial resistance alone does not explain the emergence and persistence of all shigellosis epidemics among GBMSM. In this study, we showed the previous epidemic S. sonnei strain (t10.377) was replaced by another strain of S. sonnei (t10.1814) with the same genotypic antimicrobial resistance profile, and the reemergent strain of S. flexneri 3a in 2019 was more susceptible to antimicrobials than the strain that caused the original S. flexneri 3a epidemic (41). Asymptomatic transmission of Shigella spp. among GBMSM might be a factor driving antimicrobial pressure in this group (42). Other strains during previous epidemics are examples of the emergence and persistence of strains exhibiting the same antimicrobial resistance profiles. Other factors could be at play, such as transient host immunity to circulating serotypes providing emergent serotypes with a competitive advantage. Host immunity seems an unlikely explanation for the strain replacement event because both strains were S. sonnei.
Overall, the case characteristics in the t10.1814 cluster were similar to those in the t10.377 cluster in terms of the proportion of male cases and age distribution. Some regional variation exists; cases in the t10.1814 cluster were more dispersed across regions of England, and the t10.377 cluster was more concentrated in London (Appendix Table 2). The difference in travel history between cases in the clusters could be because of missing data on recent travel history.
Phylogenetic analyses showed clustering of blaCTX-M variants within the Shigella spp. population structure, consistent with horizontal acquisition and vertical transmission. Non-GBMSM clades associated with the blaCTX-M-15 gene variant comprised cases reporting travel to high-risk regions outside the United Kingdom, highlighting the possibility that this resistance determinant was brought in through travel, similar to Shigella in other regions (43,44). One GBMSM blaCTX-M-15 gene variant isolate fell within the same 10 SNP SLC, and although the blaCTX-M-27 gene variant decrease coincided with the blaCTX-M-15 gene variant increase, there was no evidence the blaCTX-M-15 gene variant emerged from the clade with the blaCTX-M-27 gene variant. The acquisition of the blaCTX-M-15 gene variant appears to be an independent evolutionary event on a different branch of the phylogeny.
Long-read sequencing analysis revealed that, like the blaCTX-M-27 gene variant in the t10.377 cluster, the blaCTX-M-15 gene variant in the current epidemic t10.1814 cluster was located on an IncFII plasmid. Despite encoding different blaCTX-M variants, the plasmid encoding the blaCTX-M-15 gene variant exhibited high levels of similarity to the plasmid encoding the blaCTX-M-27 gene variant. Those data reveal similar IncFII plasmids persist and remain stable in the strains of S. sonnei circulating among GBMSM, despite acquisition of different antimicrobial resistance determinants. Because of the apparent plasmid stability in this population, our demonstration of the acquisition of the blaCTX-M-27 and blaCTX-M-15 gene variants and subsequent clonal expansion, the potential other antimicrobial resistance determinants could be acquired onto this plasmid and worsen the already concerning antimicrobial resistance picture of S. sonnei remains. In addition, we report an isolate in a separate clade (t10.2404) in which the blaCTX-M-15 gene variant was located on the chromosome and the associated plasmid was lost.
Social distancing and travel restrictions in 2020 and 2021 related to the COVID-19 pandemic had a greater effect on reducing notifications of S. sonnei than S. flexneri (25). Previously, we considered that globalization and increased travel might have a role in seeding sexually transmissible shigellosis. The acquisition of the blaCTX-M-15 gene variant previously associated with travel-related cases of S. sonnei, on the GBMSM-associated IncFII pKSR-100-like plasmid, may provide further evidence for this hypothesis. The reporting of S. sonnei with the blaCTX-M-15 gene variant among GBMSM in other countries in Europe suggests the potential international distribution of this lineage (45,46).
With a lack of information about sexual orientation and incomplete travel histories, it is possible that adult male case-patients who traveled were categorized as presumptive MSM within this cluster if the travel histories were not known. Identifying as GBMSM and reporting recent travel are also not mutually exclusive; therefore, there are limitations with the use of the presumptive MSM proxy definition. It is also not mandatory for primary diagnostic laboratories to send S. sonnei isolates to the gastrointestinal bacterial reference unit, so the data available for this analysis represents about two thirds of the total number of reported infections.
Despite those limitations, the introduction of WGS for typing gastrointestinal pathogens greatly improved surveillance of S. sonnei at UKHSA. Previously, we relied on phenotypic methods that were highly specialized, labor intensive, and difficult to standardize, such as phage typing and antimicrobial susceptibility testing. During the past decade, sequencing data has been used to construct the population structure of S. sonnei from UK residents and mapped clades associated with travel and associated with sexual transmission among GBMSM. We have tracked the rise and fall of different clades circulating within GBMSM sexual networks and showed that acquisition of antimicrobial resistance and genetic factors contribute to emergence, transmission, and persistence. However, notifications continue to rise, and the circulating strains are increasingly resistant to first- and second-line antimicrobial drugs.
The results in this article highlight the continued utility of genomic surveillance in detecting outbreaks of sexually transmissible shigellosis and the ever-growing importance of antimicrobial stewardship for shigellosis (47). Furthermore, through detailed analyses of the data, we can clarify the complex origins and transmission pathways for antimicrobial resistance in increasingly antimicrobial-resistant strains. We recurrently see conjugative plasmids carrying resistance against key antimicrobial classes mobilizing among Shigella spp. strains circulating in different transmission networks. This plasmid mobilization underlines the need to address Shigella spp. as an urgent antimicrobial threat, in line with the World Health Organization priority pathogen list of 2024 (48), and highlights the need to create innovative solutions to slow sexual transmission in networks in which heavy antimicrobial use drives the emergence of XDR strains.
Hannah Charles is a principal epidemiologist at the United Kingdom Health Security Agency. Her research interests include the real-time and enhanced surveillance of sexually transmissible infections, including outbreaks and incidents of Shigella.
References
- Kotloff KL, Riddle MS, Platts-Mills JA, Pavlinac P, Zaidi AKM. Shigellosis. Lancet. 2018;391:801–12. DOIPubMedGoogle Scholar
- Williamson DA, Chen MY. Emerging and reemerging sexually transmitted infections. N Engl J Med. 2020;382:2023–32. DOIPubMedGoogle Scholar
- Warren BR, Parish ME, Schneider KR. Shigella as a foodborne pathogen and current methods for detection in food. Crit Rev Food Sci Nutr. 2006;46:551–67. DOIPubMedGoogle Scholar
- Qiu S, Liu K, Yang C, Xiang Y, Min K, Zhu K, et al. A Shigella sonnei clone with extensive drug resistance associated with waterborne outbreaks in China. Nat Commun. 2022;13:7365. DOIPubMedGoogle Scholar
- Lόpez-Vélez R, Lebens M, Bundy L, Barriga J, Steffen R. Bacterial travellers’ diarrhoea: A narrative review of literature published over the past 10 years. Travel Med Infect Dis. 2022;47:
102293 . DOIPubMedGoogle Scholar - McLarty K, Paranthaman K, Jenkins C, Sedgwick J, Crawley-Boevey E. Lessons learned from the investigation and management of an outbreak of Shigella flexneri associated with a restaurant in London, 2019-2020. Public Health. 2022;205:130–2. DOIPubMedGoogle Scholar
- Mattison CP, Calderwood LE, Marsh ZA, Wikswo ME, Balachandran N, Kambhampati AK, et al. Childcare and school acute gastroenteritis outbreaks: 2009–2020. Pediatrics. 2022;150:
e2021056002 . DOIPubMedGoogle Scholar - Ryan MJ, Wall PG, Adak GK, Evans HS, Cowden JM. Outbreaks of infectious intestinal disease in residential institutions in England and Wales 1992-1994. J Infect. 1997;34:49–54. DOIPubMedGoogle Scholar
- Farrar WE Jr, Eidson M. Antibiotic resistance in Shigella mediated by R factors. J Infect Dis. 1971;123:477–84. DOIPubMedGoogle Scholar
- Davies JR, Farrant WN, Tomlinson AJ. Further studies on the antibiotic resistance of Shigella sonnei. II. The acquisition of transferable antibiotic resistance in vivo. J Hyg (Lond). 1968;66:479–87. DOIPubMedGoogle Scholar
- Dritz SK, Ainsworth TE, Back A, Boucher LA, Garrard WF, Palmer RD, et al. Patterns of sexually transmitted enteric diseases in a city. Lancet. 1977;2:3–4. DOIPubMedGoogle Scholar
- Baker KS, Dallman TJ, Ashton PM, Day M, Hughes G, Crook PD, et al. Intercontinental dissemination of azithromycin-resistant shigellosis through sexual transmission: a cross-sectional study. Lancet Infect Dis. 2015;15:913–21. DOIPubMedGoogle Scholar
- Chung The H, Rabaa MA, Pham Thanh D, De Lappe N, Cormican M, Valcanis M, et al. South Asia as a reservoir for the global spread of ciprofloxacin-resistant Shigella sonnei: a cross-sectional study. PLoS Med. 2016;13:
e1002055 . DOIPubMedGoogle Scholar - Chung The H, Boinett C, Pham Thanh D, Jenkins C, Weill FX, Howden BP, et al. Dissecting the molecular evolution of fluoroquinolone-resistant Shigella sonnei. Nat Commun. 2019;10:4828. DOIPubMedGoogle Scholar
- Baker KS, Dallman TJ, Field N, Childs T, Mitchell H, Day M, et al. Genomic epidemiology of Shigella in the United Kingdom shows transmission of pathogen sublineages and determinants of antimicrobial resistance. Sci Rep. 2018;8:7389. DOIPubMedGoogle Scholar
- Bardsley M, Jenkins C, Mitchell HD, Mikhail AFW, Baker KS, Foster K, et al. Persistent transmission of shigellosis in England is associated with a recently emerged multidrug-resistant strain of Shigella sonnei. J Clin Microbiol. 2020;58:e01692–19. DOIPubMedGoogle Scholar
- Simms I, Field N, Jenkins C, Childs T, Gilbart VL, Dallman TJ, et al. Intensified shigellosis epidemic associated with sexual transmission in men who have sex with men—Shigella flexneri and S. sonnei in England, 2004 to end of February 2015. Euro Surveill. 2015;20:21097. DOIPubMedGoogle Scholar
- Mook P, McCormick J, Bains M, Cowley LA, Chattaway MA, Jenkins C, et al. ESBL-producing and macrolide-resistant Shigella sonnei infections among men who have sex with men, England, 2015. Emerg Infect Dis. 2016;22:1948–52. DOIPubMedGoogle Scholar
- Borg ML, Modi A, Tostmann A, Gobin M, Cartwright J, Quigley C, et al. Ongoing outbreak of Shigella flexneri serotype 3a in men who have sex with men in England and Wales, data from 2009-2011. Euro Surveill. 2012;17:20137. DOIPubMedGoogle Scholar
- Ian Simms HC. Gauri Godbole, Claire Jenkins, Katy Sinka. Sexually transmitted Shigella spp. in England: 2016 to 2023. UK Health Security Agency. 2024 May 16 [cited 2024 Oct 10]. https://www.gov.uk/government/publications/non-travel-associated-shigella-infections/sexually-transmitted-shigella-spp-in-england-2016-to-2023
- Charles H, Prochazka M, Thorley K, Crewdson A, Greig DR, Jenkins C, et al.; Outbreak Control Team. Outbreak of sexually transmitted, extensively drug-resistant Shigella sonnei in the UK, 2021-22: a descriptive epidemiological study. Lancet Infect Dis. 2022;22:1503–10. DOIPubMedGoogle Scholar
- Thorley K, Charles H, Greig DR, Prochazka M, Mason LCE, Baker KS, et al. Emergence of extensively drug-resistant and multidrug-resistant Shigella flexneri serotype 2a associated with sexual transmission among gay, bisexual, and other men who have sex with men, in England: a descriptive epidemiological study. Lancet Infect Dis. 2023;23:732–9. DOIPubMedGoogle Scholar
- Lefèvre S, Njamkepo E, Feldman S, Ruckly C, Carle I, Lejay-Collin M, et al. Rapid emergence of extensively drug-resistant Shigella sonnei in France. Nat Commun. 2023;14:462. DOIPubMedGoogle Scholar
- Charles H, Sinka K, Simms I, Baker KS, Godbole G, Jenkins C. Trends in shigellosis notifications in England, January 2016 to March 2023. Epidemiol Infect. 2024;152:
e115 . DOIPubMedGoogle Scholar - Dallman TJ, Charles H, Prochazka M, Sinka K, Hughes G, Godbole G, et al. Emergence of novel strains of Shigella flexneri associated with sexual transmission in adult men in England, 2019-2020. J Med Microbiol. 2021;70:
001437 . DOIPubMedGoogle Scholar - Mitchell HD, Mikhail AFW, Painset A, Dallman TJ, Jenkins C, Thomson NR, et al. Use of whole-genome sequencing to identify clusters of Shigella flexneri associated with sexual transmission in men who have sex with men in England: a validation study using linked behavioural data. Microb Genom. 2019;5:
e000311 . DOIPubMedGoogle Scholar - Dallman T, Ashton P, Schafer U, Jironkin A, Painset A, Shaaban S, et al. SnapperDB: a database solution for routine sequencing analysis of bacterial isolates. Bioinformatics. 2018;34:3028–9. DOIPubMedGoogle Scholar
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:
e15 . DOIPubMedGoogle Scholar - Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. DOIPubMedGoogle Scholar
- Yara DA, Greig DR, Gally DL, Dallman TJ, Jenkins C. Comparison of Shiga toxin-encoding bacteriophages in highly pathogenic strains of Shiga toxin-producing Escherichia coli O157:H7 in the UK. Microb Genom. 2020;6:
e000334 . DOIPubMedGoogle Scholar - Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–6. DOIPubMedGoogle Scholar
- Wick RR, Holt KE. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLOS Comput Biol. 2022;18:
e1009802 . DOIPubMedGoogle Scholar - Hunt M, Silva ND, Otto TD, Parkhill J, Keane JA, Harris SR. Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015;16:294. DOIPubMedGoogle Scholar
- Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–903. DOIPubMedGoogle Scholar
- Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–24. DOIPubMedGoogle Scholar
- Gilchrist CLM, Chooi YH. clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics. 2021;37:2473–5. DOIPubMedGoogle Scholar
- Hawkey J, Paranagama K, Baker KS, Bengtsson RJ, Weill FX, Thomson NR, et al. Global population structure and genotyping framework for genomic surveillance of the major dysentery pathogen, Shigella sonnei. Nat Commun. 2021;12:2684. DOIPubMedGoogle Scholar
- Chowdhury G, Ghosh D, Zhou Y, Deb AK, Mukhopadhyay AK, Dutta S, et al. Field evaluation of a simple and rapid diagnostic test, RLDT to detect Shigella and enterotoxigenic E. coli in Indian children. Sci Rep. 2024;14:8816. DOIPubMedGoogle Scholar
- Lindsay B, Ochieng JB, Ikumapayi UN, Toure A, Ahmed D, Li S, et al. Quantitative PCR for detection of Shigella improves ascertainment of Shigella burden in children with moderate-to-severe diarrhea in low-income countries. J Clin Microbiol. 2013;51:1740–6. DOIPubMedGoogle Scholar
- Bardsley M, Jenkins C, Mitchell HD, Mikhail AFW, Baker KS, Foster K, et al. Persistent transmission of shigellosis in England is associated with a recently emerged multidrug-resistant strain of Shigella sonnei. J Clin Microbiol. 2020;58:e01692–19. DOIPubMedGoogle Scholar
- Mason LCE, Charles H, Thorley K, Chong CE, De Silva PM, Jenkins C, et al. The re-emergence of sexually transmissible multidrug resistant Shigella flexneri 3a, England, United Kingdom. NPJ Antimicrob Resist. 2024;2:20. DOIPubMedGoogle Scholar
- Richardson D, Savary-Trathen A, Fitzpatrick C, Williams D. Estimated prevalence and associations of sexually transmissible bacterial enteric pathogens in asymptomatic men who have sex with men: a systematic review and meta-analysis. Sex Transm Infect. 2024;100:532–7. DOIPubMedGoogle Scholar
- Kim S, Park AK, Kim JS, Park J, Shin E, Jung HJ, et al. The role of international travellers in the spread of CTX-M-15-producing Shigella sonnei in the Republic of Korea. J Glob Antimicrob Resist. 2019;18:298–303. DOIPubMedGoogle Scholar
- Campos-Madueno EI, Bernasconi OJ, Moser AI, Keller PM, Luzzaro F, Maffioli C, et al. Rapid increase of CTX-M-producing Shigella sonnei isolates in Switzerland due to spread of common plasmids and international clones. Antimicrob Agents Chemother. 2020;64:e01057–20. DOIPubMedGoogle Scholar
- Vecilla DF, Urrutikoetxea Gutiérrez MJ, Nieto Toboso MC, Inchaurza KZ, Zárraga EU, Estévez BR, et al. First report of Shigella sonnei carrying a blaCTX-M-15 sexually transmitted among men who have sex with men. Infection. 2025;53:443–8. DOIPubMedGoogle Scholar
- European Centre for Disease Prevention and Control. Communicable disease threats report: week 51, 17–23 December 2023 [cited 2024 Oct 10]. https://www.ecdc.europa.eu/sites/default/files/documents/communicable-disease-threats-report-week-51-2023.pdf
- Richardson D, Pakianathan M, Ewens M, Mitchell H, Mohammed H, Wiseman E, et al. British Association of Sexual Health and HIV (BASHH) United Kingdom national guideline for the management of sexually transmitted enteric infections 2023. Int J STD AIDS. 2023;34:588–602. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: June 17, 2025
Table of Contents – Volume 31, Number 7—July 2025
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Hannah Charles, United Kingdom Health Security Agency, 61 Colindale Ave., London NW9 5EQ, UK
Top