Volume 32, Number 4—April 2026
Dispatch
Panton-Valentine Leukocidin–Encoding Methicillin-Resistant Staphylococcus aureus, the Netherlands, 2023–20241
Figure 2
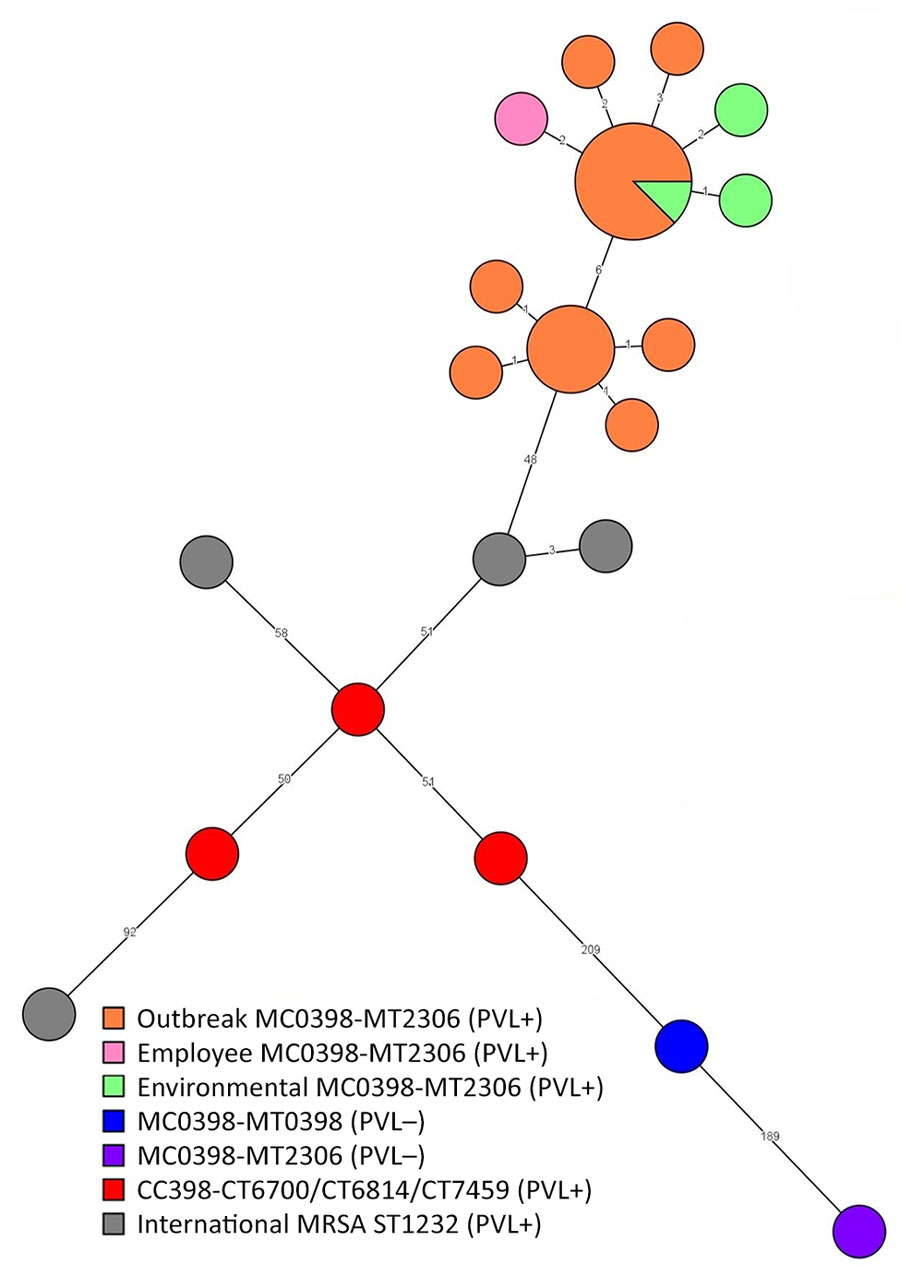
Figure 2. Whole-genome multilocus sequence typing minimum spanning tree in study of outbreak of PVL-encoding MRSA, the Netherlands, 2023–2024. We determined that all MRSA isolates were MLVA type MT2306, complex MC0398, similar to CC398. Tree shows the outbreak isolates of human cases (orange), employee case (pink), and environmental samples (green) clustering with a maximum difference of 12 alleles, confirming a microbiological link between the patients and the environmental samples. National PVL-positive, CC398 MRSA strains (red) and international PVL-positive MRSA ST1232 strains (gray) differ >48 alleles compared with the outbreak strain. The genome from a PVL-negative MC0398-MT2306 (purple) isolate from 2019 differs on 238 alleles from the outbreak isolates; the genome of the PVL-negative MC0398-MT0398 (blue) isolate differs 226 alleles from the outbreak isolates. CC, clonal complex; MLVA, multilocus variable number tandem repeat analysis; MRSA, methicillin-resistant Staphylococcus aureus; PVL, Panton-Valentine leucocidin; ST, sequence type.
1Preliminary results from this study were presented at the European Scientific Conference on Applied Infectious Disease Epidemiology (ESCAIDE); Stockholm, Sweden; November 20–22, 2024.