Volume 32, Number 7—July 2026
Research Letter
Chikungunya Outbreak, Cuba, July 2025
Figure
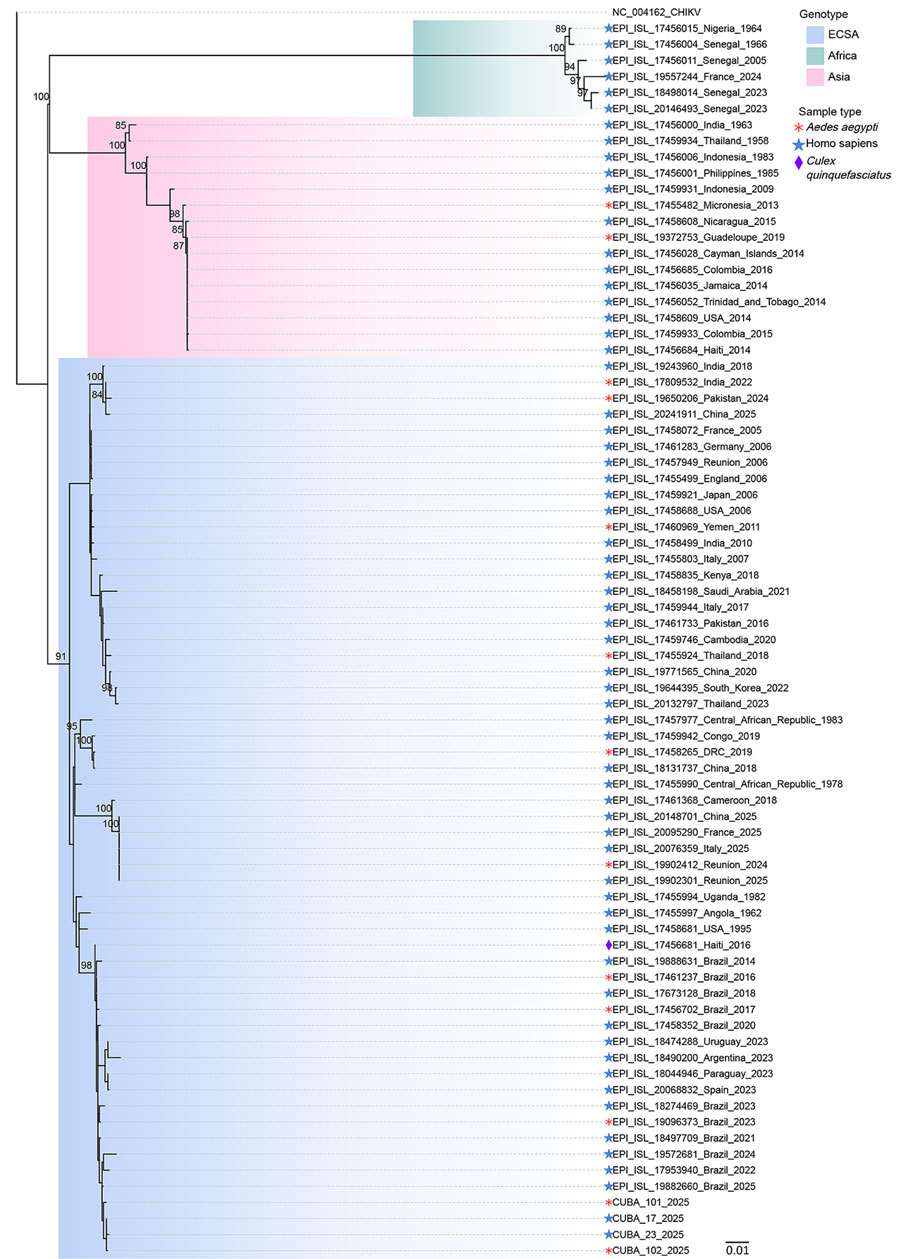
Figure. Molecular phylogenetic analysis of chikungunya virus from outbreak in Matanzas Province, Cuba, 2025. We obtained all available chikungunya virus sequences from GISAID, then filtered the sequences to ensure dataset and genetic diversity. We inferred evolutionary history by maximum-likelihood method by using the Tamura-Nei model (6). We applied the neighbor-joining method to a matrix of estimated pairwise distances to obtain initial trees and used a discrete gamma distribution to model evolutionary rate differences among sites. We conducted evolutionary analyses in MEGA version 6 (7). Numbers at branches indicate the percentage of trees in which associated taxa clustered together. Scale bar indicates number of substitutions per site. DRC, Democratic Republic of the Congo; USA, United States.
References
- Pan American Health Organization. Chikungunya: analysis by country. 2025 [cited 2025 Dec 5]. https://www.paho.org/es/arbo-portal/chikunguna-datos-analisis/chikunguna-analisis-por-pais
- de Souza WM, Ribeiro GS, de Lima STS, de Jesus R, Moreira FRR, Whittaker C, et al. Chikungunya: a decade of burden in the Americas. Lancet Reg Health Am. 2024;30:
100673 . DOIPubMedGoogle Scholar - Guzmán MG, Vázquez S, Álvarez M, Pelegrino JL, Amores DR, Martínez PA, et al. Laboratory surveillance of dengue and other arboviruses in Cuba, 1970–2017 [in Spanish]. Rev Cubana Med Trop. 2019;71:1–31.
- Naveca FG, Nascimento VAD, Souza VC, Nunes BTD, Rodrigues DSG, Vasconcelos PFDC. Multiplexed reverse transcription real-time polymerase chain reaction for simultaneous detection of Mayaro, Oropouche, and Oropouche-like viruses. Mem Inst Oswaldo Cruz. 2017;112:510–3. DOIPubMedGoogle Scholar
- General Directorate of Public Health of Havana. MINSAP. Cuban chikungunya protocol. 2025 [cited 2026 Mar 10]. https://temas.sld.cu/chikungunya/files/2025/12/protocolo-cubano-chikungunya-1.3.pdf
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20. DOIPubMedGoogle Scholar
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. DOIPubMedGoogle Scholar
- Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007;3:
e201 . DOIPubMedGoogle Scholar
1These authors contributed equally to this article.