Volume 4, Number 1—March 1998
Synopsis
What Makes Cryptococcus neoformans a Pathogen?
Cite This Article
Citation for Media
Abstract
Life-threatening infections caused by the encapsulated fungal pathogen Cryptococcus neoformans have been increasing steadily over the past 10 years because of the onset of AIDS and the expanded use of immunosuppressive drugs. Intricate host-organism interactions make the full understanding of pathogenicity and virulence of C. neoformans difficult. We discuss the current knowledge of the characteristics C. neoformans must possess to enter the host and establish progressive disease: basic growth requirements and virulence factors, such as the polysaccharide capsule; shed products of the organism; melanin production; mannitol secretion; superoxide dismutase; proteases; and phospholipases.
Figure 1
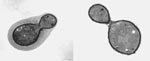
Figure 1. Transmission electron micrograph C. neoformans showing the characteristic polysaccharide capsule.
Cryptococcus neoformans is an encapsulated fungal organism (Figure 1) that can cause disease in apparently immunocompetent, as well as immunocompromised, hosts (1,2). Most susceptible to infection are patients with T-cell deficiencies (1,2). C. neoformans var. neoformans causes most cryptococcal infections in humans, so this review will focus on information from the neoformans variety of this basidiomycetous fungus. C. neoformans var. neoformans is found worldwide; its main habitats are debris around pigeon roosts and soil contaminated with decaying pigeon or chicken droppings (1,3). Not part of the normal microbial flora of humans, C. neoformans is only transiently isolated from persons with no pathologic features (2,4). It is generally accepted that the organism enters the host by the respiratory route in the form of a dehydrated haploid yeast or as basidiospores. After some time in the lungs, the organism hematogenously spreads to extrapulmonary tissues; since it has a predilection for the brain, infected persons usually contract meningoencephalitis (1). If untreated, cryptococcal meningoencephalitis is 100% fatal, and even when treated with the most effective antifungal drugs, cryptococcal infections can be fatal if the host does not have adequate T-celldependent immune function (2).
To be classified as a pathogen, an organism must be able to cause infection under certain conditions. By this definition, C. neoformans can certainly be classified as a pathogen. Because the immunodeficient are more susceptible than the immunocompetent to infection with this yeast-like organism, C. neoformans is frequently referred to as an opportunistic pathogen. The factors that make C. neoformans a pathogen can be divided into two major groups. The first comprises the basic characteristics needed to establish an infection and survive in the human host; the second comprises the virulence factors that affect the degree of pathogenicity.
The Infectious Particle
Figure 2

Figure 2. Proposed means of infection by C. neoformans.
To enter the alveolar spaces of the lungs and establish pulmonary infection, an organism must produce viable forms smaller than 4 µm in diameter. The typical vegetative form of C. neoformans is the yeast form with a cell diameter of 2.5 µm to 10 µm. The organism can also undergo sexual reproduction, and since it is a basidiomycete (Filobasidiella neoformans), it forms basidiospores. Sexual reproduction appears to occur much less frequently in nature than asexual or vegetative reproduction. The sexual spores (basidiospores) are approximately 1.8 µm to 3 µm in diameter and result from crosses of the α- and a-mating types on an appropriate medium (1). Although the exact nature of the infectious particles of C. neoformans is not known, they are presumed to be the dehydrated yeast cells or basidiospores of the appropriate size range to get into the lungs. Once inside the lungs, the yeast cells become rehydrated and acquire the characteristic polysaccharide capsule (Figure 2). In the case of basidiospores, they would convert to encapsulated blastoconidia.
Recently, Wickes et al. (5) reported that the α-mating type of C. neoformans can produce monokaryotic hyphae on a solid medium without a nitrogen source or water. The monokaryotic hyphae contain clamp connections and basidia with short chains of basidiospores and are similar in all respects except in nuclei number to the dikaryotic hyphae of the sexual forms (5). The haploid fruiting bodies formed by nitrogen-starved α-mating type isolates also produce abundant amounts of blastoconidia, which on the appropriate medium can produce haploid hyphae (5). None of the a-mating type strains tested produced the haploid fruiting bodies. Backcross studies indicated that the ability to undergo haploid fruiting lies within (or is tightly linked to) the MATα locus, which is responsible for the -mating type phenotype (5). These results suggest basidiospores from the monokaryotic hyphae may be another source of the infectious particle in nature. Regardless of the nature of the infectious particle (yeast cell or basidiospore), if a cryptococcal isolate is not able to produce small infectious particles, it cannot be pathogenic under the usual conditions for establishing the disease.
Mating Types
Kwon-Chung and Bennett (6) surveyed the mating types of C. neoformans isolates from environmental and clinical sources and found a 30- to 40-fold higher frequency of α-mating type than α-mating type. The skewed ratio of α-mating type to a-mating type was not thought to be due to a genetic preponderance towards α-mating type progeny because crosses between two test strains resulted in a 1:1 ratio of α-mating and -mating type progeny (7). To examine the reason for the disparate ratio, Kwon-Chung and colleagues (8) constructed a pair of congenic strains of C. neoformans that differed only in their mating type. Survival studies with these congenic strains demonstrated that the α-mating type (B-4500) was significantly more virulent than the a-mating strain (B-4476) when injected intravenously into mice (8). Eighty percent of mice infected with 106 B-4500 (α-mating type) were dead within 36 days; whereas, it took 93 days for 80% of mice infected with 106 B-4476 (a-mating type) to die (8). Although the mating type locus of C. neoformans has been cloned and partially characterized, the genes or gene products that contribute to the increased virulence of α-mating type isolates are not known.
The mating type locus (MATα) cloned from an α-mating type C. neoformans isolate is 35 kb to 45 kb long (9). Moore and Edman (9) demonstrated that introducing a 2.1-kb fragment of the MATα locus into an α-mating strain isolate stimulated filament formation on starvation medium. Basidia and basidiospores were not produced on the filaments. In contrast, electroporation of an α-mating strain with the same MATα DNA fragment was ineffective in stimulating filament formation under the same growth conditions (9). Sequence analysis of the 2.1-kb fragment from the MATα locus identified a 114-bp open reading frame that encodes a 38-amino acid propheromone peptide (9). The nature and the effects of the active pheromone peptide on mating and possibly virulence of C. neoformans are unknown; consequently, how the mating type relates to virulence at the genetic level is also unknown. However, the observation of haploid fruiting by α-mating type isolates of C. neoformans on nitrogen- and water-depleted medium (9) may help explain why the α-mating type isolates from clinical specimens are more frequent than the a-mating type strains (6).
Growth In Vivo
To cause infection in humans, a C. neoformans isolate must grow at 37oC in an atmosphere of approximately 5% CO2 and at a pH of 7.3 to 7.4. To survive at 37oC, the organism must have an intact gene that encodes the C. neoformans calcineurin A catalytic subunit (10). Calcineurin is a serine-threonine specific phosphatase that is activated by Ca2+-calmodulin and is involved in stress responses in yeasts (10). Although calcineurin A mutant strains of C. neoformans can grow at 24oC, they cannot survive in vitro at 37oC, in 5% CO2, or at alkaline pH (10). Since these are similar to conditions in the host, one would predict that the calcineurin A mutant would not survive in the human host. In support of that prediction, Odom et al. have shown that such mutants are not pathogenic for immunosuppressed rabbits (10). Calcineurin A appears to be a basic requirement for C. neoformans survival in the host and consequently is a necessary factor for the pathogenicity of the organism.
Virulence factors increase the degree of pathogenicity of a microbe. C. neoformans has a number of virulence factors; generally, the virulence of an isolate cannot be attributed to any single factor, but rather it is attributed to many working in unison to cause progressive disease. As virulence factors go, those of C. neoformans would be considered low-grade. We will discuss each virulence factor separately; however, the severity of the host's disease results from a combination of several virulence factors superimposed on the host's innate and immune resistance status. The virulence factors that will be discussed are capsule, cryptococcal products, melanin production, mannitol production, and potential factors such as superoxide dismutase, proteases, phospholipase B, and lysophospholipase. The polysaccharide capsule and the soluble extracellular constituents of C. neoformans (referred to here as cryptococcal products) are probably the dominant virulence factors.
C. neoformans has a capsule composed primarily of a high molecular weight polysaccharide that has a backbone of -1,3-D-mannopyranose units with single residues of ß-D-xylopyranosyl and ß-D-glucuronopyranosyl attached (11-15). This polysaccharide is referred to as glucuronoxylomannan (GXM) (11) and has four serotypes: A and D, produced by C. neoformans var. neoformans, and B and C, produced by C. neoformans var. gattii. The evidence indicates that the capsule is a key virulence factor for C. neoformans; acapsular mutants are typically avirulent, whereas encapsulated isolates have varying degrees of virulence (20). Similar observations have been made with the CAP64 gene (21,22). Although their biochemical functions have not been defined, it appears that the two genes are essential for capsule formation and virulence.
Chemotactic Effects on Leukocytes
Some properties of the C. neoformans capsule enable the host to more effectively clear the organism from tissues; however, others protect the organism from host defenses. The capsules of serotype A and D isolates are chemotactic for neutrophils (23). Moreover, complement is fixed by cryptococcal capsules by the alternative pathway (24), and this process produces chemotactic peptides such as C5a (23). Chemotaxis of leukocytes induced by either of these mechanisms would be advantageous to the host.
Effects of Complement Interactions
Complement fixing by C. neoformans in tissue would result in chemotactic factor production and attraction of leukocytes into the infection site. Once in the infected tissue, the leukocytes would interact with and kill the organism. As complement is fixed, C3b and C3bi are deposited on the surface of the cryptococci (24). The capsule can mask the C3b and C3bi deposits (24-26); however, if they are not completely masked, the deposited complement components facilitate binding of the cryptococci to CR3 receptors on leukocytes (27). Such binding interactions are advantageous to the host; they enhance the opportunity for the leukocytes to kill the cryptococci either extracellularly or after phagocytosis. The organism can also be opsonized by antibodies to GXM, but the capsule may block the Fc portion of the antibody and prevent it from binding to Fc receptors on the phagocytic host cells (28). Some of these functions of the capsule favor the host; however, if the capsule is very large, the organism is protected (24-26). The cryptococci could deplete complement in the host, creating an environment that favors the cryptococci (29).
Effects on Phagocytosis
All considered, the capsule is more beneficial to the organism than to the host. Encapsulated C. neoformans cells are not phagocytized or killed by neutrophils, monocytes, or macrophages to the same degree as acapsular mutants (25,30-33). Encapsulated C. neoformans have a stronger negatively charged surface than acapsular cells or Saccharomyces cerevisiae cells (34). The high negative charge could cause electrostatic repulsion between the organism and the negatively charged host effector cells and reduce intimate cell-cell interactions required for clearance of the cryptococci (34).
Altered Antigen Presentation
The inability of macrophages to ingest the encapsulated organisms could also diminish antigen presentation to T cells and consequently reduce immune responses. This speculation has been experimentally demonstrated by Collins and Bancroft (35). Other studies with human alveolar macrophages have confirmed that antigen presentation is not as effective with encapsulated cryptococci as with acapsular strains (33,36). Unlike acapsular cells, encapsulated isolates cannot stimulate proliferative responses in T cells because of the reduced secretion of interleukin-1 (IL-1) by the antigen-presenting cells stimulated with the encapsulated yeasts (36).
Effects on Cytokine Production
In addition to being antiphagocytic, more resistant to killing, and less able to stimulate T-cell proliferation (25,30-33,35,36), highly encapsulated isolates of C. neoformans opsonized with a heat-labile serum component, presumably complement, do not stimulate monocytes and macrophages to produce proinflammatory cytokines such as TNFα, IL-1ß, and IL-6 as effectively as similarly opsonized nonencapsulated or weakly encapsulated isolates (37-41). The signal for induction of cytokine production can be a direct result of the attachment of the monocyte or macrophage to the acapsular cryptococci or can be an outcome of the phagocytic process, induced by acapsular cryptococci. Since the capsule blocks phagocytosis, any cytokines induced by the phagocytic process would not be induced by the encapsulated C. neoformans cells. If the cryptococcal cell wall materials must be exposed to induce cytokine production, the capsule would block the direct induction of cytokine production. Regardless of the mechanism of stimulation, the lack of production of proinflammatory cytokines could have a bearing on protection. TNFα is necessary for the induction of the protective immune response against C. neoformans (42). Consequently, the lack of or reduced production of TNFα in infections with highly encapsulated isolates of C. neoformans would prevent the induction of protective immunity, resulting in progressive disease. The roles of IL-1ß and IL-6 in protecting against C. neoformans have not been defined, but it is highly probable that the lack of these two cytokines could compromise the protective responses of the host and give cryptococci the advantage.
In contrast to the reduced TNFα, IL-1ß, and IL-6 produced by stimulating monocytes and macrophages with highly encapsulated cryptococci, IL-10 produced by these host cells increases after interaction with encapsulated strains (43). Neutralization of IL-10 with anti-IL-10 in cocultures of human peripheral blood mononuclear cells and encapsulated cryptococci increased the amounts of TNFα and IL-1ß produced, which indicates that the induction of IL-10 production by stimulating macrophages with encapsulated C. neoformans downregulates the production of the proinflammatory cytokines TNFα and IL-1ß (43). One might predict that the induction of high levels of IL-10 would also preferentially stimulate the induction of a T-helper 2 (Th2) response rather than a Th1 response in the T cells (44). Since the Th1 response is associated with protection against C. neoformans (45), increased levels of IL-10 would dampen induction of the protective immune response.
Encapsulated cryptococcal cells do not affect the different types of leukocytes in the same manner. Although encapsulated isolates do not stimulate macrophages to produce proinflammatory cytokines, they do stimulate neutrophils to produce proinflammatory cytokines and the chemotactic factor IL-8 more effectively than weakly encapsulated or acapsular organisms (46). As with the stimulation of macrophages by acapsular cryptococci to produce proinflammatory cytokines, serum is required for the encapsulated organisms to induce neutrophils to produce cytokines (46). In the case of cytokine production by neutrophils in response to encapsulated C. neoformans, it appears that the opsonization process releases a factor into the supernatant that induces the neutrophils to produce the cytokines (46). How these opposing in vitro observations with macrophages and neutrophils relate to the in vivo system or pathogenicity is yet to be determined.
Cryptococcal Products
In disseminated cryptococcosis, measurable levels of cryptococcal products are present in the body fluids of the patients (47). Although GXM is the major cryptococcal component in body fluids, it is highly probable that the organism also sheds galactoxylomannan (GalXM) and mannoproteins (MP) in vivo. This speculation is based on indirect evidence from in vivo studies and on the fact that GalXM and MP are in culture medium when the organism is grown in vitro (15,48). Cryptococcal antigens in serum or spinal fluid are diagnostic for cryptococcosis (47). Furthermore, if disseminated cryptococcosis patients have high cryptococcal antigen titers at the onset of therapy, they are less likely to respond to therapy or more likely to die before therapy is completed than patients with low cryptococcal antigen titers (49). The direct relationship of cryptococcal antigen levels in body fluids and the severity of disease suggests that the cryptococcal antigens in the host's circulatory system or spinal fluid may have adverse effects on host defenses.
Effects on Leukocyte Migration
It has been recently demonstrated in the mouse model that intravascular cryptococcal antigens inhibit the migration of leukocytes from the bloodstream into an inflammatory site (50). Intravascular cryptococcal antigens significantly reduce leukocyte infiltration into a site of acute inflammation induced by such stimuli as cryptococcal culture filtrate antigen, TNFa, or the chemotactic peptide FMLP (formylmethionyl leucyl phenylalanine) or into a delayed-type hypersensitivity reaction site induced by purified protein derivative of Mycobacterium tuberculosis or by C. neoformans antigen(s) (50). Each of the identified cryptococcal products, GXM, GalXM, and MP, when given intravenously to mice, can inhibit leukocyte migration (50). Considering that GXM is the dominant cryptococcal component in the host's circulation, one might predict that GXM is mainly responsible for the reduced leukocyte migration into inflammatory sites (50). These observations imply that pulmonary cryptococcosis patients, who have low to no cryptococcal antigen concentrations in their serum, would have a normal influx of leukocytes into pulmonary sites of infections. On the other hand, in severe pulmonary infections or in disseminated cryptococcosis patients who have high levels of circulating cryptococcal antigens, minimal inflammation would be expected in the infected tissues. In fact, for years investigators have commented on the minimal host tissue responses observed in patients with disseminated cryptococcosis (3). These recent data demonstrate that the circulating cryptococcal antigens are responsible, at least in part, for the lack of host tissue response.
Cryptococcal antigen(s) can potentially prevent leukocytes from migrating into an inflammatory site in two nonexclusive ways (51,52). First, GXM can stimulate neutrophils to shed L-selectin, a surface molecule necessary for the first step in neutrophil movement into tissues (51). Without L-selectin the neutrophils do not slow down and roll along the inflamed endothelial cells lining the blood vessels. With this first step in extravasation blocked, the numbers of neutrophils in infected tissues would be greatly reduced. Second, GXM and GalXM can bind to CD18, the beta chain of the adhesion molecule LFA-1, and prevent the binding of anti-CD18 to LFA-1 (52). Consequently, this binding of GXM and GalXM could prevent LFA-1 on the neutrophils from binding to the LFA-1 ligand, ICAM-1 on the inflamed endothelial cell surface. If leukocytes are inhibited from entering tissues by either or both of these mechanisms, the organism is not effectively eliminated, and the disease is more severe.
Induction of Immunomodulatory Cells
Cryptococcal antigens injected into the bloodstream of experimental animals can induce regulatory T cells, which dampen or ablate the anticryptococcal humoral as well as cell-mediated immune responses (53-71). Some clinical correlates support the concept that the antigenemia seen in disseminated cryptococcosis downmodulates the immune responses (72,73).
A long-lasting, specific immunologic unresponsiveness has been reported in persons cured of cryptococcal meningitis (72-74). Although they made antipneumococcal polysaccharide antibodies to the same degree as control volunteers after vaccination with pneumococcal polysaccharide, cured patients could not make antibodies to cryptococcal polysaccharides after vaccination with cryptococcal antigens (72,73). Henderson et al. (72,73) speculated that the intense, prolonged antigenemia associated with the cryptococcosis may account for the observed unresponsiveness. It is not unusual for patients with disseminated cryptococcosis to have depressed cell-mediated immune (CMI) responses to cryptococcal antigens (75,76). However, sufficient information is not available to determine whether the patients had a generalized defect in CMI function or had a specifically depressed CMI response because of the cryptococcal antigenemia.
Experimental animal models demonstrate convincingly that cryptococcal antigens given intravenously can induce immunoregulatory T cells that downmodulate the anticryptococcal CMI response (56-71). Serum from C. neoformans-infected mice with a cryptococcal antigen titer of 10-4 when transferred intravenously to naive mice induces regulatory T cells that specifically depress the anticryptococcal CMI response (67). Similar immunoregulatory T cells are induced by simulating the antigenemia with intravenous injection of cryptococcal culture filtrate antigen (56,57,60-63,66-69). The immunoregulatory CD4+ T cells, which appear in the lymph nodes of the mice 7 days after antigen injection, diminish the induction of T cells responsible for the anticryptococcal delayed-type hypersensitivity response and reduce the ability of the mice to clear cryptococci from tissues (56,57,60,68). A second immunoregulatory T cell, a CD8+ cell, has been found in spleens of mice (57). The CD4+ immunoregulatory cells do not alter the numbers or types of leukocytes that infiltrate an anticryptococcal delayed-type hypersensitivity reaction; however, they do have a downregulating effect on IL-2 and IFNγ production at the site (77). In contrast, the CD8+ immunoregulatory cells appear to affect the numbers of neutrophils that infiltrate the delayed-type hypersensitivity reaction site (Murphy, unpub. data). The details of the characteristics and functions of these immunoregulatory T cells have been reviewed (45). The available data strongly support the notion that cryptococcal products in the circulation induce an array of immunoregulatory T cells, which depress the anticryptococcal immune responses and protection.
More consideration should be given to the impact of high levels of cryptococcal antigen in the cerebrospinal fluid (CSF) on progression of disease. As suggested by Denning et al. (78), high cryptococcal antigen concentrations could change the osmolality of the CSF, thereby affecting its outflow and adsorption and increasing intracranial pressure. The increased pressure may cause headaches, visual loss, and early death (78). It is also possible that release of mannitol by C. neoformans contributes to increased intracranial pressure in cryptococcal meningitis patients (78).
Melanin Synthesis
One characteristic that differentiates pathogenic isolates of C. neoformans from nonpathogenic isolates and other Cryptococcus species is the organism's ability to form a brown to black pigment on a medium (such as birdseed or caffeic acid agar) that contains diphenolic compounds (1). This pigment, first described by Staib (79), is a melaninlike compound produced by C. neoformans isolates with phenoloxidase activity (80). The importance of melanin production in C. neoformans virulence was first demonstrated in the early 1980s. Rhodes, Polacheck, and Kwon-Chung (81) reported that naturally occurring C. neoformans mutants lacking melanin (Mel-) were less virulent in mice than melanin-producing strains. Others (82-84), using chemically induced mutants or isogenic pairs of C. neoformans, have confirmed and extended this observation.
Figure 3
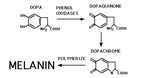
Figure 3. Proposed pathway for melanin synthesis by C. neoformans.
Biochemical analyses suggest that in C. neoformans melanogenesis is accomplished by conversion of dihydroxyphenols such as 3,4-dihydroxyphenylalanine (DOPA) to dopaquinone (Figure 3). This conversion is catalyzed by a phenoloxidase and is the rate-limiting step, presumably because subsequent steps in the pathway, such as dopaquinone rearranging to dopachrome and ultimately autoxidation to melanin, are spontaneous (85). C. neoformans lacks a tyrosinase enzyme required for endogenous production of dihydroxyphenols (83); thus to produce melanin, a C. neoformans isolate must be able to acquire diphenolic compounds from its growth environment, and it must have the enzyme phenoloxidase to catalyze conversion of these compounds into the subsequent melanin intermediates. The brain is a tissue rich in catecholamines such as DOPA and is a favorite target for infection by C. neoformans. However, the organism cannot use catecholamines as a sole carbon source, which suggests that the brain is not a preferred nutritional niche for growth of C. neoformans (86); rather, it may serve as a survival niche as described below.
Polacheck et al. (86) reported that melanin-producing isolates of C. neoformans were resistant to damage by an in vitro epinephrine oxidative system, whereas mutants lacking phenoloxidase activity were highly susceptible, as evidenced by decreased survival. Jacobson and Tinnell (87) found that melanin protected C. neoformans from damage by hypochlorite and permanganate but not by hydrogen peroxide. In these experiments, hypochlorite was 100 times more fungicidal than hydrogen peroxide, and C. neoformans could produce sufficient levels of melanin to effectively protect the organism from oxidative compounds produced by macrophages (87). Wang and Casadevall (88) extended these findings by examining survival of C. neoformans in the presence of nitric oxide and reactive nitrogen intermediates as well as in the epinephrine oxidative system described by Polacheck et al. (86). Wang and Casadevall cultured C. neoformans cells with L-DOPA to melanize the yeast cells (88). Melanized cryptococci survived damage by both nitrogen- and oxygen-derived oxidants significantly better than nonmelanized organisms of the same strain. These results support the hypothesis that C. neoformans uses catecholamines in the brain to make melanin, thereby protecting the organism from oxidative damage by scavenging free radicals (86).
Recently, cryptococcal diphenoloxidase has been purified, and the gene CNLAC1 encoding this enzyme was cloned (89). The glycosylated protein has a molecular weight of 75 kDa and contains copper. The substrate specificity of the enzyme indicates that it is a laccase. CNLAC1 contains 14 introns, a 624 amino acid open reading frame; transcriptional activity is derepressed in the absence of glucose (89), which confirms an earlier report of low glucose requirements for melanin formation (90). Disruption of CNLAC1 resulted in loss of virulence of C. neoformans, whereas complementation with CNLAC1 increased virulence of Mel- mutants in mice (84); these results suggest that the laccase (phenoloxidase) encoded by CNLAC1 is a potential virulence factor of C. neoformans. Furthermore, transcripts of the CNLAC1 gene have been detected by reverse transcription-polymerase chain reaction (RT-PCR) in C. neoformans yeast cells isolated from CSF in a rabbit model of cryptococcal meningitis (84).
Besides acting as an antioxidant, melanin production may help C. neoformans survive in the host in other ways. Melanized yeast cells are less susceptible to amphotericin B than nonmelanized yeast cells, and this may contribute to the inability to effectively treat infections in immunocompromised hosts (91). Furthermore, phagocytosis of melanized C. neoformans by a macrophage cell line in the presence of an anti-GXM antibody was decreased, which suggests that melanin deposition in the cell wall may inhibit opsonization by specific antibodies (92). Recently, Huffnagle and coinvestigators (93) reported that melanized heat-killed C. neoformans strain 145 yeast cells stimulated less TNFα production by alveolar macrophages and less antigen-specific T-cell proliferation than nonmelanized heat-killed 145 strain cells. The authors suggest that melanin "cloaks" C. neoformans from recognition by host effector cells and inhibits induction of a protective T-cell-mediated immune response (93); however, it is possible that scavenging of host-produced oxidants and inhibition of phagocytosis by melanin may contribute to the decreased TNF production and lymphoproliferation observed.
Although melanin production is important in the virulence of C. neoformans, there is very little evidence demonstrating the presence of melanin in vivo. One report indicates that phenoloxidase activity in C. neoformans is greatly diminished at 37oC compared with activity at 25oC, which suggests that melanin production may be limited in vivo (94). Detection of melanin in vivo is hampered by the lack of specific antibodies or stains. A modified Fontana-Masson stain has been used to detect a brown pigment in the cell wall of C. neoformans cells in histologic brain sections; however, the stain is not specific because Cryptococcus laurentii, which is Mel-, also stains with Fontana-Masson (95).
In summary, accumulating evidence indicates that melanin production is an effective survival (virulence) factor of C. neoformans, and melanin may serve multiple roles in protecting this medically important fungus from host defenses. Two caveats should be noted in labeling melanin as a major virulence factor for C. neoformans. The C. neoformans isolate must be able to internalize the melanin precursors, and the phenoloxidase enzyme must make a sufficient amount of melanin precursor at 37oC. Consequently, much more work is needed to delineate the role of melanin production in virulence of C. neoformans, especially in light of the apparent temperature sensitivity of the cryptococcal phenoloxidase enzyme.
Mannitol Production
Accumulating evidence suggests that production of the hexitol D-mannitol may contribute to survival of C. neoformans in the host. Wong et al. (96) reported that of the 12 human isolates of C. neoformans examined, all produced and secreted D-mannitol into growth medium. Further, by using a rabbit cryptococcal meningitis model, they showed that C. neoformans can produce D-mannitol in vivo. CSF from rabbits treated with cortisone and infected intracisternally with C. neoformans contained more D-mannitol than CSF from controls, cortisone-treated uninfected rabbits, or rabbits infected with C. neoformans but not treated with cortisone (96). In the latter group, cryptococcal infection was limited. The levels of D-mannitol in infected rabbit CSF correlated well with both the numbers of culturable C. neoformans and the cryptococcal antigen titer of the CSF, which suggests that levels of D-mannitol in CSF may be prognostic (96); however, it is not known whether different isolates of C. neoformans vary in mannitol production. These authors suggested two means by which mannitol production may contribute to C. neoformans pathogenesis. First, high concentrations of D-mannitol in the CNS may contribute to brain edema. Second, mannitol is a potent scavenger of hydroxyl radicals, and cryptococcal-produced D-mannitol may help protect the organism from oxidative damage (96).
To investigate the role of mannitol in cryptococcosis, Chaturvedi and co-workers (97) isolated a low mannitol producing mutant after UV irradiation of C. neoformans strain H99. The mutant, mannitol low producer (MLP), was similar to the parent strain H99 in many growth and morphology characteristics and in known virulence factors such as capsule production and phenoloxidase activity (97). However, the mutant MLP strain was more susceptible to growth inhibition and killing by heat and high salt than the parent strain. In addition, the mutant MLP strain was significantly less virulent than the parent strain (MLP LD50 = 3.7 X 106 CFU, H99 LD50 = 6.9 X 102) (97). Further comparisons of H99 and MLP strains showed that polymorphonuclear leukocytes (PMNL) killed MLP significantly better than H99 at several effector-to-target ratios (98). Addition of superoxide dismutase, mannitol, or dimethyl sulfoxide inhibited PMNL killing of both strains, but addition of catalase did not alter killing, which suggests that reactive oxygen intermediates such as the hydroxyl radical and hypochlorous acid are potent effector molecules against C. neoformans and mannitol may protect against oxidative killing by scavenging such compounds (98).
The results above indicate that mannitol production by C. neoformans correlates with increased resistance to heat stress, osmotic stress, and damage by reactive oxygen intermediates, as well as increased pathogenicity of this fungal agent. Additional studies are required to determine the role of mannitol production in virulence of C. neoformans. Isogenic strains lacking the enzyme required for mannitol production, namely mannitol dehydrogenase, are not available because the gene for this enzyme has not been found. However, a gene (Mtl) has been isolated from C. neoformans that can induce expression of the S. cerevisiae mannitol dehydrogenase gene (99). The 346 amino acid product encoded by the Mtl gene is thought to be involved in regulating mannitol production in C. neoformans and may be beneficial in future studies of mannitol production (99).
Superoxide Dismutase
Jacobson and coinvestigators (100) examined superoxide dismutase (SOD) production by C. neoformans to determine whether SOD levels increased at 37oC to compensate for possible decreases in melanin production at this temperature. These investigators observed an increase in SOD levels at 37oC, which suggests that SOD may participate in free radical scavenging at this higher temperature in vivo (100); however, there is no evidence that SOD production serves as a virulence factor for C. neoformans.
Proteases
Muller and Sethi (101) reported that C. neoformans grown in culture produced a protease that could digest human plasma proteins, and Brueske (102) reported that C. neoformans culture supernatants contained a protease capable of digesting casein. However, neither of these studies determined whether the proteases were manufactured in vivo. Limited evidence indicating in vivo production of proteases has been presented by Salkowski and Balish (103). These investigators observed skin lesions on T-cell—deficient mice after intravenous infections with C. neoformans strain SLHA (103). Microscopic examination of the lesions suggested that the cryptococcal yeast cells were degrading collagen bundles in the dermis (103). Supernatants of SLHA strain cultures liquefied gelatin in vitro indicating that this cryptococcal strain secreted a collagenaselike protein (103). Thus, C. neoformans proteases possibly serve as virulence mechanisms by initiating invasion of host tissues; however, more studies with isogenic strains of C. neoformans are required before proteases can be listed as virulence factors.
Phospholipases
Recently, Chen and co-workers (104,105) reported phospholipase, lysophospholipase, and lysophospholipase-transacylase activity of C. neoformans grown on egg yolk agar. Of 50 strains tested, 49 had phospholipase activity (104), due to phospholipase B secreted into cultures (105). Analysis of four strains with varying levels of phospholipase activity indicated a correlation between phospholipase activity and virulence in BALB/c mice (104). The authors suggest that extracellular phospholipase activity produced by C. neoformans may disrupt mammalian cell membranes and allow the yeast cells to penetrate into host tissues (104,105); however, further investigations are necessary to establish the role, if any, of these types of products in the virulence of C. neoformans.
Regulation of Virulence
Regulation of virulence factors such as capsule production and melanin formation is not well understood. However, the gene GPA1, which encodes a G-protein α-subunit, is involved in the regulation of these virulence factors as well as in C. neoformans mating (106). Disruption of GPA1 resulted in defects in mating in response to nitrogen starvation, capsule production in response to iron limitation, and melanin synthesis in response to glucose starvation (106). Furthermore, gpa1 mutants were much less virulent in a rabbit model of cryptococcal meningitis (106). Reconstitution of the gpa1 mutant with wild-type GPA1 restored mating, capsule production, melanin synthesis, and virulence. Addition of cyclic AMP also restored these phenotypes, which suggests that C. neoformans GPA1 regulates these factors by sensing the nutritional signals of the environment and regulating cyclic AMP metabolism in the organism (106). These findings along with results of other molecular studies are intriguing and represent the initial steps in defining at the molecular level the factors controlling virulence in C. neoformans.
Virulent isolates of C. neoformans must be able to produce small particles that can get into the alveolar spaces, must be able to grow at 37oC at a pH of 7.3 to 7.4 in an atmosphere of approximately 5% CO2, must have an intact calcineurin pathway, and (possibly) must be an -mating type. The ability to produce a large capsule and shed great amounts of capsular material into the body fluids makes the organism highly virulent. Other factors, such as melanin, mannitol, superoxide dismutase, protease, and phospholipase production, may enhance the pathogenicity of C. neoformans. The effectiveness of many of these cryptococcal virulence factors depends on the status of the host's defensive mechanisms. Although we have learned much about the pathogenicity and virulence of C. neoformans, many gaps still remain in our knowledge.
Dr. Buchanan is a research assistant professor in the Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma, USA. Dr. Murphy is a George Lynn Cross Research Professor of Microbiology and Immunology in the same department.
Acknowledgment
We thank Drs. J.A. Alspaugh, J.R. Perfect, and J. Heitman for making their manuscript available before publication.
References
- Kwon-Chung KJ. Cryptococcosis. In: Kwon-Chung KJ, Bennett JE, editors. Medical mycology. Philadelphia: Lea & Febiger, 1992:397-446.
- Mitchell TG, Perfect JR. Cryptococcosis in the era of AIDS—100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev. 1995;8:515–48.PubMedGoogle Scholar
- Rippon JW. Cryptococcosis. In: Rippon JW, editor. Medical mycology. Philadelphia: W.B. Saunders Company; 1988, p. 582-609.
- Duperval B, Hermans PE, Brewer NS, Roberts GD. Cryptococcosis, with emphasis on the significance of isolation of Cryptococcus neoformans from the respiratory tract. Chest. 1977;72:13–9. DOIPubMedGoogle Scholar
- Wickes BL, Mayorga ME, Edman U, Edman JC. Dimorphism and haploid fruiting in Cryptococcus neoformans: association with the a-mating type. Proc Natl Acad Sci U S A. 1996;93:7327–31. DOIPubMedGoogle Scholar
- Kwon-Chung KJ, Bennett JE. Distribution of and mating types of Cryptococcus neoformans among natural and clinical isolates. Am J Epidemiol. 1978;108:337–40.PubMedGoogle Scholar
- Kwon-Chung KJ, Hill WB. Sexuality and pathogenicity of Filobasidiella neoformans (Cryptococcus neoformans). In: Vanbreuseghem R, DeVroey C, Vanbreuseghem R, editors. Sexuality and pathogenicity of fungi. Paris: Masson; 181. p. 243-50.
- Kwon-Chung KJ, Edman JC, Wickes BL. Genetic association of mating types and virulence in Cryptococcus neoformans. Infect Immun. 1992;60:602–5.PubMedGoogle Scholar
- Moore TDE, Edman JC. The -mating type locus of Cryptococcus neoformans contains a pheromone gene. Mol Cell Biol. 1993;13:1962–70.PubMedGoogle Scholar
- Odom A, Muir S, Lim E, Toffaletti DL, Perfect J, Heitman J. Calcineurin is required for virulence of Cryptococcus neoformans. EMBO J. 1997;16:2576–89. DOIPubMedGoogle Scholar
- Cherniak R, Reiss E, Slodki ME, Plattner RD, Blumer SO. Structure and antigenic activity of the capsular polysaccharide of Cryptococcus neoformans. Mol Immunol. 1980;17:1025–32. DOIPubMedGoogle Scholar
- Bhattacharjee AK, Kwon-Chung KJ, Glaudemans CP. Capsular polysaccharides from a parent strain and from a possible mutant strain of Cryptococcus neoformans serotype A. Carbohydr Res. 1981;95:237–48. DOIPubMedGoogle Scholar
- Bhattacharjee AK, Bennett JE, Glaudemans CP. Capsular polysaccharides of Cryptococcus neoformans. Rev Infect Dis. 1984;6:619–24.PubMedGoogle Scholar
- Cherniak R. Soluble polysaccharides of Cryptococcus neoformans. Curr Top Med Mycol. 1988;2:40–54.PubMedGoogle Scholar
- Cherniak R, Sundstrom JB. Polysaccharide antigens of the capsule of Cryptococcus neoformans. Infect Immun. 1994;62:1507–12.PubMedGoogle Scholar
- Bulmer GS, Sans MD, Gunn CM. Cryptococcus neoformans. I. Nonencapsulated mutants. J Bacteriol. 1967;94:1475–9.PubMedGoogle Scholar
- Fromtling RA, Shadomy HJ, Jacobson ES. Decreased virulence in stable, acapsular mutants of Cryptococcus neoformans. Mycopathologia. 1982;79:23–9. DOIPubMedGoogle Scholar
- Kozel TR, Cazin J Jr. Non-encapsulated variant of Cryptococcus neoformans. I. Virulence studies and characterization of soluble polysaccharide. Infect Immun. 1971;3:287–94.PubMedGoogle Scholar
- Kwon-Chung KJ, Rhodes JC. Encapsulation and melanin formation as indicators of virulence in Cryptococcus neoformans. Infect Immun. 1986;51:218–23.PubMedGoogle Scholar
- Chang YC, Kwon-Chung KJ. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol Cell Biol. 1994;14:4912–9.PubMedGoogle Scholar
- Chang YC, Penoyer LA, Kwon-Chung KJ. The second capsule gene of Cryptococcus neoformans, CAP64, is essential for virulence. Infect Immun. 1996;64:1977–83.PubMedGoogle Scholar
- Chang YC, Cherniak R, Kozel TR, Granger LC, Morris LC, Weinhold LC, Structure and biological activities of acapsular Cryptococcus neoformans 602 complemented with the CAP64 gene. Infect Immun. 1997;65:1584–92.PubMedGoogle Scholar
- Dong ZM, Murphy JW. Mobility of human neutrophils in response to Cryptococcus neoformans cells, culture filtrate antigen, and individual components of the antigen. Infect Immun. 1993;61:5067–77.PubMedGoogle Scholar
- Kozel TR, Pfrommer GS. Activation of the complement system by Cryptococcus neoformans leads to binding of iC3b to the yeast. Infect Immun. 1986;52:1–5.PubMedGoogle Scholar
- Kozel TR, Mastroianni RP. Inhibition of phagocytosis by cryptococcal polysaccharide: dissociation of the attachment and ingestion phases of phagocytosis. Infect Immun. 1976;14:62–7.PubMedGoogle Scholar
- Kozel TR, Highison B, Stratton CJ. Localization on encapsulated Cryptococcus neoformans of serum components opsonic for phagocytosis by macrophages and neutrophils. Infect Immun. 1984;43:574–9.PubMedGoogle Scholar
- Griffin FMJ. Roles of macrophage Fc and C3b receptors in phagocytosis of immunologically coated Cryptococcus neoformans. Proc Natl Acad Sci U S A. 1981;78:3853–7. DOIPubMedGoogle Scholar
- Kozel TR, Pfrommer GS, Guerlain AS, Highison BA, Highison GJ. Role of the capsule in phagocytosis of Cryptococcus neoformans. Rev Infect Dis. 1988;10:S436–9.PubMedGoogle Scholar
- Macher AM, Bennett JE, Gadek JE, Frank MM. Complement depletion in cryptococcal sepsis. J Immunol. 1978;120:1686–90.PubMedGoogle Scholar
- Bulmer GS, Sans MD. Cryptococcus neoformans II. Phagocytosis by human leukocytes. J Bacteriol. 1967;94:1480–3.PubMedGoogle Scholar
- Bulmer GS, Sans MD. Cryptococcus neoformans. III. Inhibition of phagocytosis. J Bacteriol. 1968;95:5–8.PubMedGoogle Scholar
- Kozel TR, Gotschlich EC. The capsule of Cryptococcus neoformans passively inhibits phagocytosis of the yeast by macrophages. J Immunol. 1982;129:1675–80.PubMedGoogle Scholar
- Vecchiarelli A, Pietrella D, Dottorini M, Monari C, Retini C, Todisco T, Encapsulation of Cryptococcus neoformans regulates fungicidal activity and the antigen presentation process in human alveolar macrophages. Clin Exp Immunol. 1994;98:217–23.PubMedGoogle Scholar
- Nosanchuk JD, Casadevall A. Cellular charge of Cryptococcus neoformans: Contributions from the capsular polysaccharide, melanin, and monoclonal antibody binding. Infect Immun. 1997;65:1836–41.PubMedGoogle Scholar
- Collins HL, Bancroft GJ. Encapsulation of Cryptococcus neoformans impairs antigen-specific T-cell responses. Infect Immun. 1991;59:3883–8.PubMedGoogle Scholar
- Vecchiarelli A, Dottorini M, Pietrella D, Vecchiarelli A, Dottorini M, Pietrella D, Role of human alveolar macrophages as antigen-presenting cells in Cryptococcus neoformans infection. Am J Respir Cell Mol Biol. 1994;11:130–7.PubMedGoogle Scholar
- Levitz SM, Tabuni A, Kornfeld H, Reardon CC, Golenbock DT. Production of tumor necrosis factor alpha in human leukocytes stimulated by Cryptococcus neoformans. Infect Immun. 1994;62:1975–81.PubMedGoogle Scholar
- Naslund PK, Miller WC, Granger DL. Cryptococcus neoformans fails to induce nitric oxide synthase in primed murine macrophage-like cells. Infect Immun. 1995;63:1298–304.PubMedGoogle Scholar
- Vecchiarelli A, Retini C, Pietrella D, Monari C, Tascini T, Beccari T, Downregulation by cryptococcal polysaccharide of tumor necrosis factor alpha and interleukin-1 beta secretion from human monocytes. Infect Immun. 1995;63:2919–23.PubMedGoogle Scholar
- Delfino D, Cianci L, Migliardo M, Mancuso G, Cusumano V, Corradini C, Tumor necrosis factor-inducing activities of Cryptococcus neoformans components. Infect Immun. 1996;64:5199–204.PubMedGoogle Scholar
- Delfino D, Cianci L, Lupis E, Celese A, Petrelli ML, Curro F, Interleukin-6 production by human monocytes stimulated with Cryptococcus neoformans components. Infect Immun. 1997;65:2454–6.PubMedGoogle Scholar
- Huffnagle GB, Toews GB, Burdick MD, Boyd MB, McAllister KS, McDonald RA, Afferent phase production of TNF- required for the development of protective T cell immunity to Cryptococcus neoformans. J Immunol. 1996;157:4529–36.PubMedGoogle Scholar
- Vecchiarelli A, Retini C, Monari C, Tascini C, Bistoni F, Kozel TR. Purified capsular polysaccharide of Cryptococcus neoformans induces interleukin-10 secretion by human monocytes. Infect Immun. 1996;64:2846–9.PubMedGoogle Scholar
- Moore KW, O'Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Annu Rev Immunol. 1993;11:165–90. DOIPubMedGoogle Scholar
- Murphy JW. Cell-mediated immunity. In: Howard DH, Miller JD, editors. The Mycota VI. Human and animal relationships. Berlin: Springer-Verlag; 1996. p. 67-97.
- Retini C, Vecchiarelli A, Monari C, Tascini C, Bistoni F, Kozel TR. Capsular polysaccharide of Cryptococcus neoformans induces proinflammatory cytokine release by human neutrophils. Infect Immun. 1996;64:2897–903.PubMedGoogle Scholar
- Gordon MA, Vedder DK. Serologic tests in diagnosis and prognosis of cryptococcosis. JAMA. 1966;197:961–7. DOIPubMedGoogle Scholar
- Reiss E, Cherniak R, Eby E, Kaufman L. Enzyme immunoassay detection of IgM to galactoxylomannan of Cryptococcus neoformans. Diagn Immunol. 1984;2:109–15.PubMedGoogle Scholar
- Diamond RD, Bennett JE. Prognostic factors in cryptococcal meningitis. A study in 111 cases. Ann Intern Med. 1974;80:176–81.PubMedGoogle Scholar
- Dong ZM, Murphy JW. Intravascular cryptococcal culture filtrate (CneF) and its major component glucuronoxylomannan (GXM) are potent inhibitors of leukocyte accumulation. Infect Immun. 1995;63:770–8.PubMedGoogle Scholar
- Dong ZM, Murphy JW. Cryptococcal polysaccharides induce L-selectin shedding and tumor necrosis factor receptor loss from the surface of human neutrophils. J Clin Invest. 1996;97:689–98. DOIPubMedGoogle Scholar
- Dong ZM, Murphy JW. Cryptococcal polysaccharides bind to CD18 on human neutrophils. Infect Immun. 1997;65:557–63.PubMedGoogle Scholar
- Kozel TR, Gulley WF, Cazin J Jr. Immune response to Cryptococcus neoformans soluble polysaccharide: immunological unresponsiveness. Infect Immun. 1977;18:701–7.PubMedGoogle Scholar
- Breen JF, Lee IC, Vogel FR, Friedman H. Cryptococcal capsular polysaccharide-induced modulation of murine immune responses. Infect Immun. 1982;36:47–51.PubMedGoogle Scholar
- Murphy JW, Moorhead JW. Regulation of cell-mediated immunity in cryptococcosis. I. Induction of specific afferent T suppressor cells by cryptococcal antigen. J Immunol. 1982;128:276–83.PubMedGoogle Scholar
- Murphy JW, Mosley RL, Moorhead JW. Regulation of cell-mediated immunity in cryptococcosis. II. Characterization of first-order T suppressor cells (Ts1) and induction of second-order suppressor cell. J Immunol. 1983;130:2876–81.PubMedGoogle Scholar
- Morgan MA, Blackstock R, Bulmer GS, Hall NK. Modification of macrophage phagocytosis in murine cryptococcosis. Infect Immun. 1983;40:493–500.PubMedGoogle Scholar
- Blackstock R, Hall NK. Nonspecific immune suppression by Cryptococcus neoformans infection. Mycopathologia. 1984;86:35–43. DOIPubMedGoogle Scholar
- Murphy JW. Effects of first-order Cryptococcus-specific T-suppressor cells on induction of cells responsible for delayed-type hypersensitivity. Infect Immun. 1985;48:439–45.PubMedGoogle Scholar
- Murphy JW, Mosley RL. Regulation of cell-mediated immunity in cryptococcosis. III. Characterization of second-order T suppressor cells (Ts2). J Immunol. 1985;134:577–84.PubMedGoogle Scholar
- Mosley RL, Murphy JW, Cox RA. Immunoadsorption of Cryptococcus-specific suppressor T-cell factors. Infect Immun. 1986;51:844–50.PubMedGoogle Scholar
- Khakpour FR, Murphy JW. Characterization of a third-order suppressor T cell (Ts3) induced by cryptococcal antigen(s). Infect Immun. 1987;55:1657–62.PubMedGoogle Scholar
- Blackstock R, McCormack JM, Hall NK. Induction of a macrophage-suppressive lymphokine by soluble cryptococcal antigens and its association with models of immunological tolerance. Infect Immun. 1987;55:233–9.PubMedGoogle Scholar
- Blackstock R, Hernandez NC. Inhibition of phagocytosis in cryptococcosis: phenotypic analysis of the suppressor cell. Cell Immunol. 1988;114:174–87. DOIPubMedGoogle Scholar
- Murphy JW, Cox RA. Induction of antigen-specific suppression by circulating Cryptococcus neoformans antigen. Clin Exp Immunol. 1988;73:174–80.PubMedGoogle Scholar
- Murphy JW. Influence of cryptococcal antigens on cell-mediated immunity (CMI). Rev Infect Dis. 1988;10:5432–5.
- Murphy JW. Clearance of Cryptococcus neoformans from immunologically suppressed mice. Infect Immun. 1989;57:1946–52.PubMedGoogle Scholar
- Murphy JW. Immunoregulation in cryptococcosis. In: Kurstak E, editor. Immunology of fungal diseases. New York: Marcel Dekker; 1989. p. 319-45.
- Blackstock R, Hall NK, Hernandez NC. Characterization of a suppressor factor that regulates macrophage phagocytosis in murine cryptococcosis. Infect Immun. 1989;57:1773–9.PubMedGoogle Scholar
- Blackstock R, Zembala M, Asherson GL. Functional equivalence of cryptococcal and haptene-specific T suppressor factor (TsF). I. Picryl and oxazolone-specific TsF, which inhibit transfer of contact sensitivity also inhibit phagocytosis by a subset of macrophages. Cell Immunol. 1991;136:435–47. DOIPubMedGoogle Scholar
- Henderson DK, Bennett JE, Huber MA. Long-lasting, specific immunologic unresponsiveness associated with cryptococcal meningitis. J Clin Invest. 1982;69:1185–90. DOIPubMedGoogle Scholar
- Henderson DK, Kan VL, Bennett JE. Tolerance to cryptococcal polysaccharide in cured cryptococcosis patients: failure of antibody secretion in vitro. Clin Exp Immunol. 1986;65:639–46.PubMedGoogle Scholar
- Salvin SB, Smith RF. An antigen for detection of hypersensitivity to Cryptococcus neoformans. Proc Soc Exp Biol Med. 1961;108:498–501.PubMedGoogle Scholar
- Diamond RD, Bennett JE. Disseminated cryptococcosis in man: decreased lymphocyte transformation in response to Cryptococcus neoformans. J Infect Dis. 1973;127:694–7.PubMedGoogle Scholar
- Graybill JR, Alford RH. Cell-mediated immunity in cryptococcosis. Cell Immunol. 1974;14:12–21. DOIPubMedGoogle Scholar
- Buchanan KL, Murphy JW. Regulation of cytokine production during the expression phase of the anticryptococcal delayed-type hypersensitivity response. Infect Immun. 1994;62:2930–9.PubMedGoogle Scholar
- Denning DW, Armstrong RW, Lewis BH, Stevens DA. Elevated cerebrospinal fluid pressures in patients with cryptococcal meningitis and acquired immunodeficiency syndrome. Am J Med. 1991;91:267–72. DOIPubMedGoogle Scholar
- Staib F. Cryptococcus neoformans und Guizotia abyssinica. Zeitschrift für Hygiene. 1962;148:466–75. DOIGoogle Scholar
- Shaw CE, Kapica L. Production of diagnostic pigment by phenoloxidase activity of Cryptococcus neoformans. Appl Microbiol. 1972;24:824–30.PubMedGoogle Scholar
- Rhodes JC, Polacheck I, Kwon-Chung KJ. Phenoloxidase activity and virulence in isogenic strains of Cryptococcus neoformans. Infect Immun. 1982;36:1175–84.PubMedGoogle Scholar
- Kwon-Chung KJ, Polacheck I, Popkin TJ. Melanin-lacking mutants of Cryptococcus neoformans and their virulence for mice. J Bacteriol. 1982;150:1414–21.PubMedGoogle Scholar
- Torres-Guerrero H, Edman JC. Melanin-deficient mutants of Cryptococcus neoformans. J Med Vet Mycol. 1994;32:303–13. DOIPubMedGoogle Scholar
- Salas SD, Bennett JE, Kwon-Chung KJ, Perfect JR, Williamson PR. Effect of the laccase gene, CNLAC1, on virulence of Cryptococcus neoformans. J Exp Med. 1996;184:377–86. DOIPubMedGoogle Scholar
- Polacheck I, Kwon-Chung KJ. Melanogenesis in Cryptococcus neoformans. J Gen Microbiol. 1988;134:1037–41.PubMedGoogle Scholar
- Polacheck I, Platt Y, Aronovitch J. Catecholamines and virulence of Cryptococcus neoformans. Infect Immun. 1990;58:2919–22.PubMedGoogle Scholar
- Jacobson ES, Tinnell SB. Antioxidant function of fungal melanin. J Bacteriol. 1993;175:7102–4.PubMedGoogle Scholar
- Wang Y, Casadevall A. Susceptibility of melanized and nonmelanized Cryptococcus neoformans to nitrogen- and oxygen-derived oxidants. Infect Immun. 1994;62:3004–7.PubMedGoogle Scholar
- Williamson PR. Biochemical and molecular characterization of the diphenol oxidase of Cryptococcus neoformans: identification as a laccase. J Bacteriol. 1994;176:656–64.PubMedGoogle Scholar
- Polacheck I, Hearing VJ, Kwon-Chung KJ. Biochemical studies of phenoloxidase and utilization of catecholamines in Cryptococcus neoformans. J Bacteriol. 1982;150:1212–20.PubMedGoogle Scholar
- Wang Y, Casadevall A. Growth of Cryptococcus neoformans in presence of L-dopa decreases its susceptibility to amphotericin B. Antimicrob Agents Chemother. 1994;38:2648–50.PubMedGoogle Scholar
- Wang Y, Aisen P, Casadevall A. Cryptococcus neoformans melanin and virulence: mechanism of action. Infect Immun. 1995;63:3131–6.PubMedGoogle Scholar
- Huffnagle GB, Chen GH, Curtis JL, McDonald RA, Strieter RM, Toews GB. Down-regulation of the afferent phase of T cell-mediated pulmonary inflammation and immunity by a high melanin-producing strain of Cryptococcus neoformans. J Immunol. 1995;155:3507–16.PubMedGoogle Scholar
- Jacobson ES, Emery HS. Temperature regulation of the cryptococcal phenoloxidase. J Med Vet Mycol. 1991;29:121–4. DOIPubMedGoogle Scholar
- Kwon-Chung KJ, Hill WB, Bennett JE. New, special stain for histopathological diagnosis of cryptococcosis. J Clin Microbiol. 1981;13:383–7.PubMedGoogle Scholar
- Wong B, Perfect JR, Beggs S, Wright KA. Production of the hexitol D-mannitol by Cryptococcus neoformans in vitro and in rabbits with experimental meningitis. Infect Immun. 1990;58:1664–70.PubMedGoogle Scholar
- Chaturvedi V, Flynn T, Niehaus WG, Wong B. Stress tolerance and pathogenic potential of a mannitol mutant of Cryptococcus neoformans. Microbiology. 1996;142:937–43. DOIPubMedGoogle Scholar
- Chaturvedi V, Wong B, Newman SL. Oxidative killing of Cryptococcus neoformans by human neutrophils. Evidence that fungal mannitol protects by scavenging reactive oxygen intermediates. J Immunol. 1996;156:3836–40.PubMedGoogle Scholar
- Perfect JR, Rude TH, Wong B, Flynn T, Chaturvedi V, Niehaus W. Identification of a Cryptococcus neoformans gene that directs expression of the cryptic Saccharomyces cerevisiae mannitol dehydrogenase gene. J Bacteriol. 1996;178:5257–62.PubMedGoogle Scholar
- Jacobson ES, Jenkins ND, Todd JM. Relationship between superoxide dismutase and melanin in a pathogenic fungus. Infect Immun. 1994;62:4085–6.PubMedGoogle Scholar
- Muller HE, Sethi KK. Proteolytic activity of Cryptococcus neoformans against human plasma proteins. Med Microbiol Immunol (Berl). 1972;158:129–34. DOIGoogle Scholar
- Brueske CH. Proteolytic activity of a clinical isolate of Cryptococcus neoformans. J Clin Microbiol. 1986;23:631–3.PubMedGoogle Scholar
- Salkowski CA, Balish E. Cutaneous cryptococcosis in athymic and beige-athymic mice. Infect Immun. 1991;59:1785–9.PubMedGoogle Scholar
- Chen SCA, Muller M, Zhou JZ, Wright LC, Sorrell TC. Phospholipase activity in Cryptococcus neoformans: a new virulence factor? J Infect Dis. 1997;175:414–20. DOIPubMedGoogle Scholar
- Chen SCA, Wright LC, Santangelo RT, Muller M, Moran VR, Kuchel PW, Identification of extracellular phospholipase B, lysophospholipase, and acyltransferase produced by Cryptococcus neoformans. Infect Immun. 1997;65:405–11.PubMedGoogle Scholar
- Alspaugh JA, Perfect JR, Heitman J. Cryptococcus neoformans mating and virulence are regulated by the G-protein subunit GPA1 and cAMP. Genes Dev. 1997;11:3206–17. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 4, Number 1—March 1998
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Juneann W. Murphy, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA; fax: 405-271-3117
Top