Volume 5, Number 1—February 1999
Synopsis
Comparative Genomics and Host Resistance against Infectious Diseases
Cite This Article
Citation for Media
Abstract
The large size and complexity of the human genome have limited the identification and functional characterization of components of the innate immune system that play a critical role in front-line defense against invading microorganisms. However, advances in genome analysis (including the development of comprehensive sets of informative genetic markers, improved physical mapping methods, and novel techniques for transcript identification) have reduced the obstacles to discovery of novel host resistance genes. Study of the genomic organization and content of widely divergent vertebrate species has shown a remarkable degree of evolutionary conservation and enables meaningful cross-species comparison and analysis of newly discovered genes. Application of comparative genomics to host resistance will rapidly expand our understanding of human immune defense by facilitating the translation of knowledge acquired through the study of model organisms. We review the rationale and resources for comparative genomic analysis and describe three examples of host resistance genes successfully identified by this approach.
Two major elements underlie a thorough understanding of the pathogenesis of virtually any infectious disease: identification and characterization of the virulence factors and in vivo survival mechanisms of the invading microorganism (e.g., surface attachment factors, exotoxins, or enzymes that disrupt cellular homeostasis [1]) and understanding of the components of the host response that lead to elimination of the invading pathogen and resolution of disease. (These include both nonspecific [or innate] immune defense mechanisms, such as the complement cascade, and adaptive elements, such as clonally derived lymphocytes capable of eliminating specific targets [2]). The traditional approach to human infectious diseases has been to focus research on the study of important pathogens. The outcome of investigation of relevant bacteria, viruses, fungi, and parasites has led to the production of protective vaccines, antimicrobial agents, and effective strategies for control and elimination of disease outbreaks. A principal advantage of microbiologic research is the relative ease with which the organisms may be obtained, manipulated, and analyzed in the laboratory. Because microbial genomes are smaller, complete cloning and DNA sequencing of several microorganisms have been achieved and have paved the way for comprehensive study of gene expression and genome organization (3,4). In contrast are relatively limited advances in our understanding of the molecular basis of host defense. The study of host immune defense in humans is inherently complex; obstacles to greater understanding include limited opportunities for controlled observation and experimental manipulation, a large genome, and until recently, a lack of molecular techniques capable of facilitating genomewide analysis.
One of the principal aims of the study of host response to infectious diseases is to uncover novel components of the host immune system critical to robust host defense. Identification of these components at a molecular level is the first step in understanding how the host deals with an infectious challenge and lays a foundation upon which rational therapies that augment host resistance may someday be designed. Despite this promise, the interaction between host and pathogen that leads to infection is multidimensional, dynamic, and exceedingly complex. From a genomic perspective, a thorough understanding of the pathogenesis of a given infection would include a complete inventory of the spatial and temporal expression of the genes by both the host and pathogen from the time of exposure to the final resolution of the infection. Given the potentially large number of factors that contribute to host defense, precise gene identification is a formidable challenge. Nevertheless, researchers have recently made progress in dissecting and identifying the most important individual genetic elements that govern the host response to important pathogenslargely through the use of animal models of human disease (5). Of the model organisms amenable to genetic analysis, the mouse is by far the most well-developed and physiologically relevant system for study of human host defense (6,7). Identification of commercially available inbred strains of mice that show a differential response to a well-defined infectious challenge is the first requirement for study of genetically regulated host resistance factors. Once distinct phenotypes are identified, controlled breeding is carried out to determine the mode of inheritance of the phenotype (simple or complex). Correlation of the inheritance of susceptibility or resistance to a specific infectious challenge with one or more chromosomal regions is then performed by using linkage analysis. Finally, known genes within the genetic interval must be evaluated and novel genes must be positionally cloned to elucidate the underlying molecular basis of immune defense. Comparative genomic analysis is a logical extension of these principles (8). Knowledge of the genomic organization of human and mouse, for example, facilitates direct localization and identification of the human orthologues of susceptibility genes identified through experimental challenge. These genes can then be tested as candidates for human disease susceptibility through mutation analysis.
Genetic linkage maps provide an organizational framework for genes and phenotypes in the genome (9). Maps, by establishing the location, order, and relative distance of genes, anonymous DNA markers, and biologically important traits along a species' chromosomes, are critical tools in analyzing genetic contribution to a given disease state. Genetic maps can help precisely localize chromosomal region(s) linked to host resistance phenotypes and provide the starting point for identification of the causative gene(s). During the past decade, comprehensive genetic maps spanning the genomes of mouse and human, have been created largely through the initiative of the Human Genome Project (10).
Mapping the Human Genome
A great deal of effort has resulted in the creation of a whole genome human linkage map, consisting of 5,624 microsatellite markers located to 2,335 positions (11). The DNA markers in this map are highly informative and are densely distributed, with an average interval between markers of 1.6 centimorgans (cM) (1 cM = a 1% rate of recombination during meiosis, or approximately 1 million bp). Other comprehensive maps have been assembled on the basis of a collection of more than 16,000 distinct transcribed sequences (including known genes and gene fragments) or expressed sequence tags, which are estimated to represent at least 50% of all genes in the human genome (12). This human transcription map has been integrated with selected microsatellite markers from the Généthon collection, thus allowing the position of gene-based markers to be resolved to specific intervals measured in centimorgans. The map is available electronically (13). Work is also under way to generate comprehensive physical maps of the human genome in which the relative location of markers is defined by the actual length along the chromosome, rather than by recombination events (14,15).
Mapping the Mouse Genome
Figure
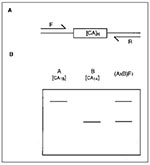
Figure. Schematic representation of microsatellite marker analysis in mice. A) Flanking forward (F) and reverse (R) oligonucleotides are designed to specifically amplify a simple sequence repeat by polymerase chain reaction (PCR) (in...
Among model organisms, genetic mapping is most well established in the mouse, having begun in 1915 with the discovery of the first linkage group (16). Controlled crosses of common laboratory strains segregating a small number of visible phenotypes such as coat color then became the mainstay of genetic mapping. In the past decade, two major breakthroughs have revolutionized the technique of mouse genetic mapping and paved the way for generation of high-resolution whole genome maps. The first was the development of the interspecific cross, involving a laboratory strain (Mus musculus) and a distantly related species Mus spretus (17), allowing literally thousands of genes to be mapped within the same cross. The second advance was the development of abundant genetic markers rapidly typable by polymerase chain reaction (PCR) (termed microsatellites), which amplified polymorphisms in simple sequence length repeats such as [CA]n (Figure) (18). Several comprehensive genetic maps of the mouse (based on genes or microsatellites) have been developed, and in some cases, these are being integrated. At least three are publicly available, while the others are available for mapping in a collaborative arrangement (19). As of January 1997, more than 17,000 markers had been mapped in the mouse (one locus approximately every 200kb), including more than 5,000 genes and more than 10,000 (mostly microsatellite) DNA markers.
Mapping in Other Species
Genetic mapping has been widely embraced by the scientific community; more than 30 vertebrate species are the subject of genetic mapping projects, and high-resolution maps of microsatellite markers have been developed for humans, mice, rats, cows, sheep, pigs, fish, and chickens (19). Two invertebrates, Drosophila melanogaster (a dipteran fly) and Caenorhabditis elegans (a nematode), also have complete genetic and physical maps; the complete nucleotide sequence of the latter is expected in the near future. The status of individual genetic mapping projects and resources has been summarized, along with a compilation of databases for species-specific or comparative mapping reference (19,20). Integrating the data from these species-specific projects in a form that allows relevant information from diverse organisms to be assembled is a major challenge to biologic information systems. The most extensive coverage of mammalian species homologies is the Mouse Genome Database of The Jackson Laboratory (21). Initially developed for the mouse, comparative mapping data for more than 55 species may be searched online, with links to related genomic resources, such as the Human Genome Database, Ratmap, SheepBase, and PigBase.
Because of the density of genetic markers positioned along the chromosomes of both organisms, the comparative map of the mouse and human genomes is the most well developed of all species. In a comprehensive summary of mouse/human homology published in 1996, 1,416 loci were placed on both maps by using human physical mapping data and mouse genetic maps (22). This comparison defined 181 conserved linkage groups, approximately 90% of the mouse genome. Further comparative mapping with newly discovered genes and expressed sequence tags will refine the chromosomal relationships between mouse and human.
The integration of existing genetic maps of different species is a formidable challenge. Accurate, comprehensive comparisons of gene arrangements across different species will rapidly advance our understanding of all aspects of biology by allowing rapid information exchange across different model organisms and experimental systems. Several approaches have been used in developing universal mapping probes for diverse genomes (23-25). Of the two classes of loci used to construct gene maps, coding gene sequences (Type I markers), which show conservation among distantly related mammalian species, are most useful as landmarks for comparing linkage and syntenic association. Highly polymorphic sequences (Type II markers), such as microsatellites, are more abundant and are invaluable for mapping within a pedigree but are less useful for comparative purposes because they do not show adequate sequence conservation to recognize locus homology between mammalian orders. In 1993, a list of anchored reference loci for comparative genome mapping in mammals was proposed; it consists of 321 Type I markers equivalently spaced throughout the mammalian genome (26). This approach allowed the position of homologous loci in the maps of four species (human, mouse, cattle, and cat), which represent different mammalian orders, to be established. Interspecies comparison of conserved exon sequences of homologous genes has generated a new overlapping set of anchor loci called comparative anchor tagged sequences (25). Large-scale mapping of these sequences in several species may be an efficient way of developing high-resolution comparative maps with essentially complete genome coverage.
Mammalian genomes may be compared at several levels by using a variety of tools and strategies tailored to individual objectives. Although direct sequence comparison of whole genomes will provide the highest resolution for comparative study, this sophisticated form of analysis is at least several years away from being realized. At a cytologic level, species may be compared by fluorescence in situ hybridization (FISH) with single or multiple probes (single or multicolor Zoo-FISH), producing rapid, high-resolution chromosomal localization detectable by microscopy. Alternatively, libraries from microdissected or individual flow-sorted chromosomes may be constructed and used as fluorescence-labeled chromosome "paints" to probe the chromosomes of other species and identify homologous regions (27,28). The main advantage of chromosome painting is its rapid overall evaluation of the extent and character of genomic conservation among distantly related species, such as pig and cattle. In contrast to FISH, chromosome painting does not allow determination of gene order or high-resolution demarcation of chromosomal breakpoints. Radiation hybrid panels, another method for physical assignment of homologous loci (29,30), are generated by irradiation and subsequent fusion of a cell line containing a chromosome from one species, such as human, on another background, such as hamster. The donor DNA is fragmented at random, resulting in a series of lines retaining only fragments of the original chromosome. Conserved genes from other species may be mapped to the homologous region of the human genome by comparing the PCR pattern for each cell line to reference loci with well-established map positions.
Identifying genetically regulated host immune responses might significantly advance our understanding of the molecular targets and immunologic mechanisms critical to robust defense against pathogenic microbes. To date the number of host defense genes that have been cloned remains small; comparative genomics has the potential to accelerate gene discovery by allowing available data for model organisms to be rapidly applied to the study of human disease. We summarize three examples of human host resistance genes in the following section; in each example, genetic analysis of mouse models of the human disease phenotype played a crucial role in the initial discovery of the human homologue or served as a means of validating the identity of the proposed human candidate disease gene.
The Mouse Nramp1 Gene
In classic inbred strains of mice, natural resistance to infection with Mycobacterium bovis (BCG), M. lepraemurium, Salmonella Typhimurium, and Leishmania donovani is controlled by the Bcg locus, also known as Ity and Lsh (31-33). The major effect of the Bcg gene is to modulate the growth rate of these diverse pathogens in cells of the reticuloendothelial system of the mouse during the preimmune phase of the infection (33). Resistant and susceptible strains are distinguished by the kinetics of infection shown by pathogen counts (CFUs or Leishmania-forming units) in liver and spleen after infection. The susceptible phenotype is characterized by a higher net growth rate of BCG, Salmonella, or Leishmania in the reticuloendothelial system during the early phase of infection, followed by specific immune responses in BCG- and L. donovaniinfected mice or by a rapidly lethal infection with the virulent pathogen S. Typhimurium. Bcg is inherited as a simple autosomal dominant Mendelian trait in crosses between classical strains of laboratory mice; it was localized to mouse chromosome 1 by linkage analysis (34). Using a positional cloning strategy, Vidal et al. (35) isolated the Nramp1 (natural resistance-associated macrophage protein 1) gene as a strong candidate for the Bcg mutation based on its map location, its macrophage-restricted expression pattern and a nonconservative Gly169Asp substitution in the protein of all susceptible strains. Creation of a null allele at Nramp1 then provided formal proof that a mutation within Nramp1 is the cause of the mouse susceptibility to infection with M. bovis, S. Typhimurium, and L. donovani (36).
Nramp1, an integral membrane phosphoglycoprotein located in the late endosome/lysosome compartment of resting macrophages, is recruited to the maturing phagosomal membrane (37), consistent with its potential function in controlling the replication of intracellular parasites by altering the intravacuolar environment in which they reside. Nramp1 is part of an ancient family of proteins with highly conserved members in mammals (including humans, cows, rats, sheep), birds, invertebrates (C. elegans, D. melanogaster), plants (Oryza sativa, Arabidopsis thaliana), fungi (Saccharomyces cerevisiae), and even bacteria (M. leprae and Escherichia coli) (38,39). This family is characterized by a highly conserved hydrophobic core consisting of 10 transmembrane (TM) domains with a structural organization typical of families of ion transporters and channels. In addition, the most highly conserved segments of the Nramp family (TM8-TM9 intracellular loop) show impressive similarity with the highly conserved region of mammalian voltage-gated K+ channels of the shaker type (40).
Several issues concerning the biochemical function of Nramp1 with respect to intracellular survival of taxonomically unrelated pathogens remain unresolved. Studies of the function of Nramp1-related sequences (Nramp2 and Smf1 in model organisms) provide insight into how Nramp1 confers resistance to microbial agents. Nramp2 has been isolated in mouse and human and shows a high degree of similarity to Nramp1 (77% overall similarity), with identical hydropathy profiles and predicted secondary structures (41,42). Mouse and human Nramp2 mRNA are both widely expressed in contrast with the tissue-specific expression of Nramp1 (41,42). Recently, Nramp2 was shown to be a metal ion transporter with broad divalent cation specificity (including Fe2+, Zn2+, Mn2+, Co2+, Cd2+, Cu2+, Ni2+, and Pb2+), driven by the proton electrochemical gradient in Xenopus laevis oocytes (43). Studies using the yeast double mutant SMF1/SMF2 provided additional support concerning the function of Nramp2 as a divalent cation transporter. Inactivation of SMF1 and SMF2, two yeast Nramp homologues encoding divalent cation transporters (44), is specifically complemented by Nramp2 (45). In vivo, Nramp2 plays an important role in normal iron transport. Mutation within Nramp2 causes microcytic anemia in mk mutant mice because of severe defects in intestinal iron uptake (46). Interestingly, the missense mutations in mutant Nramp1 and Nramp2 alleles introduce a charged amino acid in two adjacent positions of TM4, confirming the importance of this region of both proteins for normal function. It has been suggested that Nramp1 may also be a divalent cation transporter; its role in reticuloendothelial cells remains unexplored (40,44).
The Chicken NRAMP1 Gene
The discovery of Nramp1 allowed the study of its role in susceptibility to related infections in other species. Salmonellosis, one of the most common causes of food poisoning in humans, is frequently caused by ingestion of contaminated poultry products; efforts to identify salmonella resistance genes in poultry could lead to more efficient poultry control strategies, thereby reducing secondary human morbidity. Genetic regulation of chicken host resistance exists, as inbred poultry lines differ in their susceptibility to infection with several strains of Salmonella. Segregation analysis with a combination of Salmonella-resistant and Salmonella-susceptible lines has shown that resistance to infection is fully dominant and is not sex-linked or associated with the major histocompatibility complex (47). The candidacy of the chicken Nramp1 homologue was tested in the differential resistance of inbred chicken lines to infection with S. Typhimurium by using sequencing and linkage analyses (48). Through the use of a mouse cDNA, the chicken homologue Nramp1 has been cloned and shown to share 68% identity with the mouse gene (49). As demonstrated in mice, the macrophage is a major site of NRAMP1 mRNA expression in chickens (49). NRAMP1 mRNA transcripts from S. Typhimuriumresistant or susceptible chickens were analyzed to identify amino acid sequence variants that could be associated with the disease phenotype. Eleven sequence variants in Nramp1 mRNA were obtained from three Salmonella-resistant and three Salmonella-susceptible chicken lines; almost all (10) resulted in silent mutations or conservative changes (to amino acids with similar physical properties) that were detected both in resistant and susceptible chicken lines, while only one sequence variant resulted in a non-conservative substitution of a positively charged residue (Arg223 by a polar residue (Gln223). This allelic variant was specific to the susceptible line C and was clearly associated with survival to infection (a resistance allele at NRAMP1 improved survival rate from 13% to 27%) (48). Taken together, these data strongly suggest a direct role of NRAMP1 in susceptibility to infection in chickens.
The Human NRAMP1 Gene
Work in inbred strains of mice has established unambiguously that Nramp1 has an important role in determining resistance to mycobacterial infections and has encouraged several research groups to test the association of NRAMP1 with corresponding human infections. Host genetic factors play a major role in determining the outcome of mycobacterial infections in humans, as shown by racial variation in susceptibility to infection and higher concordance of tuberculosis and leprosy among monozygotic twins compared with dizygotic twins and siblings (50,51). Segregation analysis in a population from Desirade Island (French West Indies) has demonstrated that susceptibility to leprosy (regardless of the clinically defined subtype) is controlled by a major gene not linked to the major histocompatibility complex (52). Through use of a candidate gene approach, population association studies, and linkage analysis, several genes (HLA-linked genes, tumor necrosis factor, collectin, vitamin D receptor, interferon gamma receptor) have each been associated with susceptibility to mycobacterial infections (53,54).
The chromosomal region surrounding Nramp1 on mouse chromosome 1 has been conserved on the telomeric end of human chromosome 2q35 and contains the human NRAMP1 orthologue (55). Sequence comparison of the mouse and human Nramp1/NRAMP1 proteins showed a high degree of conservation between the two species (85% identity, 92% similarity); the most conserved region was the intracellular loop containing the consensus sequence transport motif (56). In humans, the highest sites of NRAMP1 expression are peripheral blood leukocytes and lungs (56). The high degree of sequence homology between mouse and human NRAMP1, the presence of similar regulatory elements within the promoter regions of the genes, and similar tissue expression patterns support the notion that the NRAMP1 protein exerts similar roles in vivo in both mouse and humans.
A number of polymorphic variants have been used to study the association of NRAMP1 and susceptibility to leprosy and tuberculosis (57-60). One study based on the segregation analysis of certain NRAMP1 haplotypes in 20 multiplex families involving 168 individuals from South Vietnam clearly showed that NRAMP1 was involved in predisposition to leprosy (61). Another large study measuring the association of NRAMP1 with clinical tuberculosis in a population of Gambia (West Africa) demonstrated that polymorphic variations within the human NRAMP1 gene affect susceptibility to the disease (62). Nevertheless, susceptibility to either leprosy or tuberculosis appears to be genetically heterogeneous since the role of NRAMP1 was observed only in certain ethnic groups (63,64).
Identification of Nramp1 illustrates the value of comparative genomics for identification and characterization of the biologic basis for differences between susceptible and resistant hosts. Genetic dissection of the mouse model of M. bovis infection was crucial to the identification of similar mechanisms governing the human response to medically important pathogens such as tuberculosis and leprosy. Comparative genomics was also important in accelerating the identification of an important host resistance gene for salmonellosis in the chicken (a species of significant agricultural importance), where the available genetic tools are modest, relative to mice and humans.
CHS is a rare autosomal recessive disorder characterized by partial ocular and cutaneous albinism, a mild bleeding diathesis, and peripheral sensorimotor neuropathy. The most serious phenotype among CHS patients, however, is a marked increased in susceptibility to bacterial infection that may lead to death during the first 2 decades of life. These clinical features are attributable to dysfunctional granule-containing cells including melanocytes, platelets, Schwann cells, neurons, and granulocytes (65,66). On the basis of phenotypic similarity, the beige (bg) mutation in mice has long been regarded as a model for CHS (67). Several components of the immune system are affected in Beige/CHS. Neutrophils exhibit defective chemotaxis and reduced intracellular killing for up to 90 minutes after bacterial phagocytosis, and their granules lack the serine proteases cathepsin G and elastase because of a failure of normal protein sorting (68,69). Natural killer cell activity is defective, causing impaired cytolysis of tumors and virally infected cells; cytotoxic T-cell responses against allogeneic tumor cells are also abnormal (70,71). Mice with the bg mutation have increased susceptibility to a variety of pathogens, including cytomegalovirus, Leishmania donovani, Candida albicans, and a variety of pathogenic bacteria (E. coli, Klebsiella pneumoniae, Staphylococcus aureus, Streptococcus pneumoniae) (72-74).
To identify the genetic basis of this host resistance defect, the bg gene was localized to a 0.24 cM interval of proximal mouse chromosome 13 by genetic mapping of three mouse back-crosses segregating this phenotype (75). A DNA contig of this region spanning 2,400 kb was constructed from large-capacity yeast artificial chromosomes and P1 bacteriophage clones (76). Using yeast artificial chromosome complementation and direct cDNA selection, two groups subsequently identified portions of a candidate gene for bg, named Lyst (lysosomal trafficking regulator) (77,78). Lyst, ubiquitously expressed in the mouse, has a maximum transcript size of approximately 12kb and possible complex alternative splicing. Several mutations predicted to severely truncate the Lyst polypeptide were identified within each transcript. Through the use of partial sequence data for mouse Lyst, 27 cDNAs corresponding to the human gene were identified and assembled into a complete human gene sequence of 13,499 bp, with an open reading frame of 11,403 bp (79). Comparison of the partial 3' mouse cDNA to the human sequence demonstrated 77.2% nucleotide identity and 87.9% amino acid identity, indicating that human and mouse genes are highly homologous, and sequence analysis of three CHS patients identified pathologic mutations in all.
Comparative genetic mapping between the region of mouse chromosome 13 with the bg mutation and the human genome indicates homology with distal chromosome 1q. Consistent with this alignment, genetic mapping of the human CHS locus in affected families localized it to 1q42-1q44 as part of a conserved linkage group shared with mouse chromosome 13 (80,81). Radiation hybrid mapping also assigned the human CHS candidate gene to 1q43, confirming that the bg phenotype in mouse and human CHS are both caused by mutations in orthologous genes (79). Database searches with the complete nucleotide sequence of the CHS gene showed significant homology to open reading frames from S. cerevisiae and C. elegans, as well as a human cell division control protein-4 (CDC4L) (82). The modular architecture of the CHS protein is similar to Vps15, a yeast serine/threonine kinase protein kinase thought to be part of a membrane-associated signal transduction complex regulating intracellular protein trafficking (83). To date, the function of the CHS gene remains unknown, although it may be similar to Vps15 and may be part of a novel gene family.
XLA, one of the first primary immunodeficiency disorders described in humans, is the prototypic example of the protective role of humoral immunity against common bacterial pathogens (84). XLA is characterized by a profound deficiency of B-lymphocyte development at two sequential stages of maturation within the bone marrow (85). This defect results in marked reductions in the serum levels of all three major classes of immunoglobulins and a profound decrease in the number of B lymphocytes in the peripheral blood as well as in the lymphoid follicles and germinal centers of lymph nodes. The clinical manifestations generally begin by the end of the first year of life, once the level of maternally derived antibodies has declined. Bacterial infections with organisms such as S. pneumoniae, Haemophilus influenzae, S. aureus, and Pseudomonas species are most common, with the respiratory tract being most frequently affected. Gastrointestinal infection with Salmonella or Campylobacter have also been reported, as have urogenital infections with Mycoplasma or Chlamydia. XLA patients have defective host resistance to enteroviruses, since neutralizing antibody is important in controlling these pathogens during their passage through the blood stream. Resistance to other infections for which intact T lymphocyte function is required (e.g., tuberculosis or histoplasmosis) remains intact.
Recognition of the familial occurence of this rare disorder and pedigree analysis demonstrated an X-linked recessive inheritance pattern of the trait (86). Carrier females could not be detected because they are phenotypically normal, with normal serum levels of immunoglobulin. Linkage studies of over 500 individuals from 60 families mapped the gene for XLA to the midportion (Xq22) of the X chromosome, cosegregating with the polymorphic genetic marker DXS178 (87,88). By using complementary strategies of positional cloning and low-stringency cDNA library screening, two groups identified a novel src-like cytoplasmic tyrosine kinase, named Btk (Bruton agammaglobulinemia tyrosine kinase) as a strong candidate gene for XLA (89,90). Btk was mapped to the XLA locus by FISH and somatic cell hybrid analysis and was expressed in cell lines representing all stages of B cell development, myelomonocytic cell lines, and a macrophage cell line; it was not detectable in T lineage cell lines (90). In transformed B-cell lines from individuals affected with XLA, the expression level of Btk mRNA and protein, and consequently its kinase activity, was reduced or absent. Southern blot analysis of DNA from pedigrees with XLA cases showed restriction fragment length alterations that segregated in an X-linked recessive pattern; detailed analysis disclosed either genomic DNA deletions in the region encompassing Btk or missense point mutations resulting in nonconservative amino acid substitutions at important residues in the putative protein-tyrosine kinase domain (89). These findings provide strong evidence that the failure of normal B-cell growth and differentiation in XLA is caused by abnormal function of an intracellular protein tyrosine kinase.
The CBA/N inbred mouse strain's X-linked immunodeficiency (xid) has been regarded as an experimental model for human XLA since it was first described in 1972 (91). B lymphocytes from these mice exhibit pleiotropic defects in development and function. Normal numbers of pro-B, pre-B, and surface immunoglobulin-positive B cells exist in the bone marrow, while peripheral B-cell numbers are significantly reduced (30% of normal). The B lymphocytes that are present have an abnormal surface marker phenotype, and B-cell proliferation triggered through the surface immunoglobulin M (IgM) receptor or surface immunoglobulin cross-linking is impaired, as are responses to a number of other mitogenic stimuli including lipopolysaccharide, interleukins IL-5 and IL-10, CD38 receptors, and CD40 ligands. Consistent with these defects, CBA/N mice have reduced serum IgM and IgG3 antibody levels and cannot make antibody responses when challenged with type-2 thymus-independent antigens (e.g., polysaccharides and hapten-polysaccharide conjugates). As with human XLA, impaired humoral immunity can result in increased susceptibility to bacterial pathogens, including S. Typhimurium (92). Inheritance of the susceptibility trait was linked to the xid locus by using back-cross and F2 progeny derived from crosses of CBA/N and DBA/2N parental strains.
To determine whether XLA and xid were caused by mutations in homologous genes, two groups performed genetic mapping of xid and Btk. The Btk gene was closely linked to the xid locus in the distal region of the mouse X chromosome by using an interspecific back-cross mapping panel (93), and precise co-localization of Btk and xid was observed in 1,114 segregating back-cross progeny (94). Normal and mutant mouse strains did not differ in Btk expression or in vitro kinase activity. Sequence analysis of the mouse Btk transcript in CBA/N and several immunocompetent mouse strains (including the CBA/CaHN progenitor) demonstrated a point mutation within the first coding exon that is predicted to convert a highly conserved arginine residue to cysteine. This amino acid substitution occurs within the pleckstrin homology domain in the amino-terminal region of the protein and is presumed to alter normal B-cell signaling by disrupting protein-protein interactions. To unequivocally confirm that mutations in Btk were responsible for the xid phenotype, targeted gene disruption (a gene knockout experiment) was performed in embryonic stem cells (95,96). Complete elimination of Btk protein production identically reproduced the xid phenotype, indicating that the naturally occurring point mutation produces a complete loss-of-function phenotype or results in a protein with dominant negative properties (presence of a single mutant allele is sufficient to block normal gene function). The severe early B-lymphocyte developmental arrest of human XLA was not observed, which suggests that Btk function in mice is accompanied by a compensatory mechanism operating during early B-cell development to rescue B-cell maturation.
On the basis of comparative mapping and sequence analysis, human XLA and the mouse xid phenotype are clearly homologous disorders caused by mutations in orthologous genes. Nevertheless, although the underlying genetic alteration in both species was successfully identified, a number of issues remain unresolved. First, the phenotypes observed in these two disorders are not identical; the more severe block of early lymphocyte development in XLA results in a greater deficiency of peripheral B cells relative to the CBA/N mouse strain, suggesting that the requirement for Btk in early murine B-cell development is less stringent than that for humans. Second, the range of pathogens to which humans are are highly susceptible appears more diverse than the range for mice. Finally, the exact role of Btk in normal B-cell physiology remains to be demonstrated. Thus far, identification of BTK has led to carrier detection and prenatal counselling; additional characterization of a mouse model with great similarity to the human condition could advance our understanding of the fundamental processes underlying B-lymphocyte development and function.
Complete understanding of infectious disease pathogenesis requires identification and characterization of host genes that regulate the response to virulent microorganisms. Through evolutionary selection, a series of innate immune defense mechanisms have evolved to protect the host against the constant threat of microbial injury and direct the development of specific adaptive immune responses. Genetic analysis of naturally occurring variation in the host response among model organisms has successfully identified novel genes such as Nramp1, Lyst, and Btk, thus providing new insights into the molecular nature of host resistance. Rapid advances are now being made in the creation and integration of dense genetic maps of model organisms and humans. Comparative genomics will play an increasingly important role in facilitating the transfer of new knowledge from experimental models to a more complete understanding of human host resistance.
Dr. Qureshi is a research fellow at the Centre for the Study of Host Resistance, McGill University, and assistant physician (infectious diseases) at Montréal General Hospital. His research focuses on genetic analysis of host resistance against bacterial diseases.
Acknowledgment
This work was supported by grants to Danielle Malo from the Medical Research Council of Canada (MRC), and the Canadian Bacterial Diseases Network.
References
- Finlay BB, Cossart P. Exploitation of mammalian host cell functions by bacterial pathogens. Science. 1997;276:718–25. DOIPubMedGoogle Scholar
- Tang CM, Hood DW, Moxon ER. Haemophilus influence: the impact of whole genome sequencing on microbiology. Trends Genet. 1997;13:399–404. DOIPubMedGoogle Scholar
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Life with 6,000 genes. Science. 1996;274:546–67. DOIPubMedGoogle Scholar
- Malo D, Skamene E. Genetic control of host resistance to infection. Trends Genet. 1994;10:365–71. DOIPubMedGoogle Scholar
- Bedell MA, Jenkins NA, Copeland NG. Mouse models of human disease. Part I: techniques and resources for genetic analysis in mice. Genes Dev. 1997;11:1–10. DOIPubMedGoogle Scholar
- Eppig JT, Nadeau JH. Comparative maps: the mammalian jigsaw puzzle. Curr Opin Genet Dev. 1995;5:709–16. DOIPubMedGoogle Scholar
- Copeland NG, Jenkins NA, Gilbert DJ, Eppig JT, Maltais LJ, Miller JC, A genetic linkage map of the mouse: current applications and future prospects. Science. 1993;262:57–66. DOIPubMedGoogle Scholar
- Dib C, Fauré S, Fizames C, Samson D, Druout N, Vignal A, A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–4. DOIPubMedGoogle Scholar
- Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, Rice K, A gene map of the human genome. Science. 1996;274:540–6. DOIPubMedGoogle Scholar
- National Center for Biotechnology Information. A gene map of the human genome [document on line]. Available from http://www.ncbi.nlm.nih.gov/SCIENCE96/
- Chumakov IM, Rigault P, Le Gall I, Bellanné-Chantelot C, Billault A, Guillou S, A YAC contig map of the human genome. Nature. 1995;377(Supp):175–83.PubMedGoogle Scholar
- Hudson TJ, Stein LD, Gerety SS, Ma J, Castle AB, Silva J, An STS-based map of the human genome. Science. 1995;270:1945–54. DOIPubMedGoogle Scholar
- Dietrich WF, Copeland NG, Gilbert DJ, Miller JC, Jenkins NA, Lander ES. Mapping the mouse genome: current status and future prospects. Proc Natl Acad Sci U S A. 1995;92:10849–53. DOIPubMedGoogle Scholar
- Avner P, Amar L, Dandolo L, Guénet JL. Genetic analysis of the mouse using interspecific crosses. Trends Genet. 1988;4:18–23. DOIPubMedGoogle Scholar
- Love JM, Knight AM, McAleer MA, Todd JA. Towards construction of a high resolution map of the mouse genome using PCR-analyzed microsatellites. Nucleic Acids Res. 1991;18:4123–30. DOIGoogle Scholar
- Andersson L, Archibald A, Ashburner M, Audun S, Barendse W, Bitgood J, Comparative genome organization of vertebrates. Mamm Genome. 1996;7:717–34. DOIPubMedGoogle Scholar
- Wakefield MJ, Graves JAM. Comparative maps of vertebrates. Mamm Genome. 1996;7:715–6. DOIPubMedGoogle Scholar
- Nadeau JH, Grant PL, Mankala S, Reiner AH, Richardson JE, Eppig JT. A Rosetta Stone of mammalian genetics. Nature. 1995;373:363–5. DOIPubMedGoogle Scholar
- DeBry RW, Seldin MF. Human/mouse homology relationships. Genomics. 1996;33:337–51. DOIPubMedGoogle Scholar
- Hino O, Testa JT, Buetow KH, Taguchi T, Zhou J-Y, Bremer M, Universal mapping probes and the origin of chromosome 3. Proc Natl Acad Sci U S A. 1993;90:730–4. DOIPubMedGoogle Scholar
- Mazzarella R, Montanaro V, Kere J, Reinbold R, Ciccodicola A, D'Urso M, Conserved sequence-tagged sites: a phylogenetic approach to genome mapping. Proc Natl Acad Sci U S A. 1992;89:3681–5. DOIPubMedGoogle Scholar
- Lyons LA, Laughlin TF, Copeland NG, Jenkins NA, Womack JE, O'Brien SJ. Comparative anchor tagged sequences (CATS) for integrative mapping of mammalian genomes. Nat Genet. 1997;15:47–56. DOIPubMedGoogle Scholar
- O'Brien SJ, Womack JE, Lyons LA, Moore KJ, Jenkins NA, Copeland NG. Anchored reference loci for comparative genome mapping in mammals. Nat Genet. 1993;3:103–12. DOIPubMedGoogle Scholar
- Weinberg J, Stanyon R. Comparative painting of mammalian chromosomes. Curr Opin Genet Dev. 1997;7:784–91. DOIPubMedGoogle Scholar
- Scherthan H, Cremer T, Arnason U, Weier H-U, Lima-de-Faria A, Frönicke L. Comparative chromosome painting discloses homologous segments in distantly related mammals. Nat Genet. 1994;6:342–7. DOIPubMedGoogle Scholar
- Cox DR, Burmeister M, Price ER, Kim S, Myers RM. Radiation hybrid mapping: a somatic cell genetic method for constructing high-resolution maps of mammalian chromosomes. Science. 1990;250:245–50. DOIPubMedGoogle Scholar
- Walter MA, Goodfellow PN. Radiation hybrids: irradiation and fusion gene transfer. Trends Genet. 1995;9:352–6. DOIGoogle Scholar
- Bradley DJ. Genetic control of Leishmania populations within the host. II. Genetic control of acute susceptibility of mice to L. donovani infections. Clin Exp Immunol. 1977;30:130–40.PubMedGoogle Scholar
- Plant JE, Glynn A. Genetics of resistance to infection with Salmonella typhimurium in mice. J Infect Dis. 1976;133:72–8.PubMedGoogle Scholar
- Gros P, Skamene E, Forget A. Genetic control of natural resistance to Mycobacterium bovis (BCG) in mice. J Immunol. 1981;127:2417–21.PubMedGoogle Scholar
- Skamene E, Gros P, Forget A, Kongshavn PAL, St Charles C, Taylor B. Genetic regulation of resistance to intracellular pathogens. Nature. 1982;297:506–9. DOIPubMedGoogle Scholar
- Vidal S, Malo D, Vogan K, Skamene E, Gros P. Natural resistance to infection with intracellular parasites: isolation of a candidate gene for Bcg. Cell. 1993;73:469–85. DOIPubMedGoogle Scholar
- Vidal S, Tremblay ML, Govoni G, Gauthier S, Sebastiani G, Malo D, The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J Exp Med. 1995;182:655–66. DOIPubMedGoogle Scholar
- Gruenheid S, Pinner E, Desjardins M, Gros P. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med. 1997;185:717–30. DOIPubMedGoogle Scholar
- Cellier M, Prive G, Belouchi A, Kwan T, Rodrigues V, Chia W, Nramp defines a family of membrane proteins. Proc Natl Acad Sci U S A. 1995;92:10089–93. DOIPubMedGoogle Scholar
- Hu J, Bumstead N, Burke D, Ponce de Leon FA, Skamene E, Gros P, Genetic and physical mapping of the natural resistance associated macrophage protein 1 (NRAMP1) in chicken. Mamm Genome. 1995;6:809–15. DOIPubMedGoogle Scholar
- Cellier M, Belouchi A, Gros P. Resistance to intracellular infections: comparative genomic analysis of Nramp. Trends Genet. 1996;12:201–4. DOIPubMedGoogle Scholar
- Vidal S, Belouchi AM, Cellier M, Beatty B, Gros P. Cloning and characterization of a second human NRAMP gene on chromosome 12q13. Mamm Genome. 1995;6:224–30. DOIPubMedGoogle Scholar
- Grunheid S, Cellier M, Vidal S, Gros P. Identification and characterization of a second mouse Nramp gene. Genomics. 1995;25:514–25. DOIPubMedGoogle Scholar
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romerao MF, Boron WF, Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–8. DOIPubMedGoogle Scholar
- Supek F, Supekova L, Nelson H, Nelson N. Functional of metal-ion homeostasis in the cell division cycle, mitochondrial protein processing, sensitivity to mycobacterial infection and brain function. J Exp Biol. 1997;200:321–30.PubMedGoogle Scholar
- Pinner E, Gruenheid S, Raymond M, Gros P. Functional complementation of the yeast divalent cation transporter family SMF by NRAMP2, a member of the mammalian natural resistance-associated macrophage protein family. J Biol Chem. 1997;272:28933–8. DOIPubMedGoogle Scholar
- Fleming MD, Trenor CC, Su MA, Foernzler D, Veier DR, Dietrich WF, Microcytic anemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–90.PubMedGoogle Scholar
- Bumstead N, Barrow PA. Genetics of resistance to Salmonella typhimurium in newly hatched chicks. Br Poult Sci. 1988;29:521–9. DOIPubMedGoogle Scholar
- Hu J, Bumstead N, Barrow P, Sebastiani G, Olien L, Morgan K, Resistance to salmonellosis in the chicken is linked to NRAMP1 and TNC. Genome Res. 1997;7:693–704.PubMedGoogle Scholar
- Hu J, Bumstead N, Skamene E, Gros P, Malo D. Structural organization, sequence and expression of the chicken NRAMP1 gene encoding the natural resistance associated macrophage protein 1. DNA Cell Biol. 1996;15:113–23. DOIPubMedGoogle Scholar
- Comstock GW. Tuberculosis in twins: a re-analysis of the Prophit survey. Am Rev Respir Dis. 1978;117:621–4.PubMedGoogle Scholar
- Abel L, Vu VD, Oberti J, Nguyen VT, Van VC, Guilloud-Bataille M, Complex segregation analysis of leprosy in Vietnam. Genet Epidemiol. 1995;12:63–82. DOIPubMedGoogle Scholar
- Abel L, Demenais L. Detection of major genes for susceptibility to leprosy and its subtypes in a Caribbean island: Desirade island. Am J Hum Genet. 1988;42:256–66.PubMedGoogle Scholar
- Hill AVS. Genetics of infectious disease resistance. Curr Opin Genet Dev. 1996;6:348–53. DOIPubMedGoogle Scholar
- Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile J-F, Newport M, Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N Engl J Med. 1997;335:1956–61. DOIGoogle Scholar
- Schurr E, Skamene E, Morgan K, Chu M-L, Gros P. Mapping of Col3a1 and Col6a3 to proximal murine chromosome 1 identifies conserved linkage of structural protein genes between murine chromosome 1 and human chromosome 2q. Genomics. 1990;8:477–86. DOIPubMedGoogle Scholar
- Cellier M, Govoni G, Vidal S, Kwan T, Groulx N, Liu J, Human natural resistance-associated macrophage protein: cDNA cloning, chromosomal mapping, genomic organization, and tissue-specific expression. J Exp Med. 1994;180:1741–52. DOIPubMedGoogle Scholar
- Liu J, Fujiwara TM, Buu NT, Sanchez FO, Cellier M, Paradis AJ, Identification of polymorphisms and sequence variants in the human homologue of the mouse natural resistance-associated macrophage protein gene. Am J Hum Genet. 1995;56:845–53.PubMedGoogle Scholar
- Buu NT, Cellier M, Gros P, Schurr E. Identification of a highly polymorphic length variant in the 3' UTR of NRAMP1. Immunogenetics. 1995;42:428–9. DOIPubMedGoogle Scholar
- White JK, Shaw M-A, Barton CH, Cerretti DP, Williams H, Mock BA, Genetic and physical mapping of 2q35 in the region of NRAMP and IL8R genes: identification of a polymorphic repeat in exon 2 of NRAMP. Genomics. 1994;24:295–302. DOIPubMedGoogle Scholar
- Blackwell JM. Genetics of host resistance and susceptibility to intramacrophage pathogens: a study of multicase families of tuberculosis, leprosy and leishmaniasis in north-eastern Brazil. Int J Parasitol. 1998;28:21–8. DOIPubMedGoogle Scholar
- Abel L, Sanchez FO, Oberti J, Thuc NV, Van Hoa L, Lap VD, Susceptibility to leprosy is linked to the human NRAMP1 gene. J Infect Dis. 1998;177:133–45. DOIPubMedGoogle Scholar
- Bellamy R, Ruwende C, Corrah T, McAdam KPWJ, Whittle HC, Hill AVS. Variations in the NRAMP1 gene and susceptibility to tuberculosis in west Africans. N Engl J Med. 1998;338:640–4. DOIPubMedGoogle Scholar
- Shaw MA, Atkinson S, Dockrell H, Hussain R, Lins-Lainson Z, Shaw J, An RFLP map for 2q33-q37 from multicase mycobacterial and leishmanial disease families: no evidence for an Lsh/Ity/Bcg gene homologue influencing susceptibility to leprosy. Ann Hum Genet. 1993;57:251–71. DOIPubMedGoogle Scholar
- Levee G, Liu J, Gicquel B, Chanteau S, Schurr E. Genetic control of susceptibility to leprosy in French Polynesia: no evidence for linkage with markers on telomeric human chromosome 2q. Int J Lepr Other Mycobact Dis. 1994;62:499–511.PubMedGoogle Scholar
- Malech HL, Nauseef WM. Primary inherited defects in neutrophil function: etiology and treatment. Semin Hematol. 1997;34:279–90.PubMedGoogle Scholar
- Blume RS, Wolff SM. The Chediak-Higashi syndrome: studies in four patients and a review of the literature. Medicine (Baltimore). 1972;51:247–80. DOIPubMedGoogle Scholar
- Kelley EM. Beige. Mouse News Lett. 1957;16:36.
- Gallin JI, Bujak JS, Patten E, Wolff SM. Granulocyte function in the Chediak-Higashi syndrome of mice. Blood. 1974;43:201–6.PubMedGoogle Scholar
- Ganz T, Metcalf JA, Gallin JI, Boxer LA, Lehrer RI. Microbicidal/cytotoxic proteins of neutrophils are deficient in two disorders: Chediak-Higashi syndrome and "specific" granule deficiency. J Clin Invest. 1988;82:552–6. DOIPubMedGoogle Scholar
- Saxena RK, Saxena QB, Adler WH. Defective T-cell response in beige mutant mice. Nature. 1982;295:240–1. DOIPubMedGoogle Scholar
- Haliotis T, Roder J, Klein M, Ortaldo J, Fauci A, Heberman R. Chediak-Higashi gene in humans I. Impairment of natural killer function. J Exp Med. 1980;151:1039–48. DOIPubMedGoogle Scholar
- Shellam GR, Allen JE, Papadimitrious JM, Bancroft GJ. Increased susceptibility to cytomegalovirus infection in beige mutant mice. Proc Natl Acad Sci U S A. 1981;78:5104–8. DOIPubMedGoogle Scholar
- Kirkpatrick CE, Farrell JP. Leishmaniasis in beige mice. Infect Immun. 1982;38:1208–16.PubMedGoogle Scholar
- Elin RJ, Edelin JB, Wolff SM. Infection and immunoglobulin concentrations in Chediak-Higashi mice. Infect Immun. 1974;10:88–91.PubMedGoogle Scholar
- Kingsmore SF, Barbosa MDFS, Tchernev VT, Detter JC, Lossie AC, Holcomb RF. Positional cloning of the Chediak-Higashi syndrome gene: genetic mapping of the beige locus on mouse chromosome 13. J Investig Med. 1996;44:454–61.PubMedGoogle Scholar
- Kingsmore SF, Barbosa MDFS, Nguyen QA, Ashley JA, Blaydes SM, Tchernev VT, Physical mapping of the beige critical region on mouse chromosome 13. Mamm Genome. 1996;7:773–5. DOIPubMedGoogle Scholar
- Perou CM, Moore KJ, Nagle DJ, Misumi DJ, Woolf EA, McGrail SH, Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet. 1996;13:303–8. DOIPubMedGoogle Scholar
- Barbosa MDFS, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382:262–5. DOIPubMedGoogle Scholar
- Nagle DJ, Karim MA, Woolf EA, Holmgren K, Bork P, Misumi DJ, Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14:307–11. DOIPubMedGoogle Scholar
- Fukai K, Oh J, Karim MA, Moore KJ, Kandil HH, Ito H, Homozygosity mapping of the gene for Chediak-Higashi syndrome to chromosome 1q42-q44 in a segment of conserved synteny that includes the mouse beige locus (bg). Am J Hum Genet. 1996;59:620–4.PubMedGoogle Scholar
- Barrat FJ, Auloge L, Pastural E, Dufourcq Lagelouse R, Vilmer E, Cant AJ, Genetic and physical mapping of the Chediak-Higashi syndrome on chromosome 1q42-43. Am J Hum Genet. 1996;59:625–32.PubMedGoogle Scholar
- Feuchter AE, Freeman JD, Mager DL. Strategy for detecting cellular transcripts promoted by human endogenous long terminal repeats: identification of a novel gene (CDCL4) with homology to yeast CDC4. Genomics. 1992;13:1237–46. DOIPubMedGoogle Scholar
- Klionsky DJ, Emr SD. A new class of lysosomal/vacuolar protein sorting signals. J Biol Chem. 1990;265:5349–52.PubMedGoogle Scholar
- Ochs HD, Edvard Smith CI. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine (Baltimore). 1996;75:287–99. DOIPubMedGoogle Scholar
- Kwan S-P, Kunkel L, Bruns G, Wedgewood RJ, Latt S, Rosen FS. Mapping of the X-linked agammaglobulinemia locus by use of restriction fragment-length polymorphism. J Clin Invest. 1986;77:649–52. DOIPubMedGoogle Scholar
- Guiolo S, Arveiler B, Bardoni B, Notarangelo LD, Panina P, Duse M, Close linkage of probe p212 (DXS178) to X-linked agammaglobulinemia. Hum Genet. 1989;84:19–21. DOIPubMedGoogle Scholar
- Kwan S-P, Terwilliger J, Parmley R, Raghu G, Sandkuyl LA, Ott J, Identification of a closely linked DNA marker, DXS178, to further refine the X-linked agammaglobulinemia locus. Genomics. 1990;6:238–42. DOIPubMedGoogle Scholar
- Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, The gene involved in X-linked agammaglobulinemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–33. DOIPubMedGoogle Scholar
- Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Cutler Allen R, Klisak I, Deficient expression of a B cell cytoplasmic kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–90. DOIPubMedGoogle Scholar
- Scher I. The CBA/N mouse strain: an experimental model illustrates the influence of the X-chromosome on immunity. Adv Immunol. 1982;33:1–71. DOIPubMedGoogle Scholar
- O'Brien AD, Scher I, Campbell GH, MacDermott GH, Formal SB. Susceptibility of CBA/N mice to infection with Salmonella Typhimurium: influence of the X-linked gene controlling B lymphocyte function. J Immunol. 1979;123:720–4.PubMedGoogle Scholar
- Rawlings DJ, Saffran DC, Tsukata S, Largaespada DA, Grimoldi JC, Cohen L, Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–61. DOIPubMedGoogle Scholar
- Thomas DJ, Sideras P, Edvard Smith CI, Vorechovsky I, Chapman V, Paul W. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–8. DOIPubMedGoogle Scholar
- Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–99. DOIPubMedGoogle Scholar
- Kerner JD, Appleby MW, Mohr RN, Chien S, Rawlings DJ, Maliszewski CR, Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–12. DOIPubMedGoogle Scholar
Figure
Cite This ArticleTable of Contents – Volume 5, Number 1—February 1999
| EID Search Options |
|---|
|
|
|
|
|
|