Volume 5, Number 2—April 1999
Synopsis
Bacterial Toxins: Friends or Foes?
Cite This Article
Citation for Media
Abstract
Many emerging and reemerging bacterial pathogens synthesize toxins that serve as primary virulence factors. We highlight seven bacterial toxins produced by well-established or newly emergent pathogenic microbes. These toxins, which affect eukaryotic cells by a variety of means, include Staphylococcus aureus α-toxin, Shiga toxin, cytotoxic necrotizing factor type 1, Escherichia coli heat-stable toxin, botulinum and tetanus neurotoxins, and S. aureus toxic-shock syndrome toxin. For each, we discuss the information available on its synthesis and structure, mode of action, and contribution to virulence. We also review the role certain toxins have played in unraveling signal pathways in eukaryotic cells and summarize the beneficial uses of toxins and toxoids. Our intent is to illustrate the importance of the analysis of bacterial toxins to both basic and applied sciences.
Since diphtheria toxin was isolated by Roux and Yersin in 1888 (1), microbial toxins have been recognized as the primary virulence factor(s) for a variety of pathogenic bacteria. Bacterial toxins have been defined as "soluble substances that alter the normal metabolism of host cells with deleterious effects on the host" (2). Indeed, the major symptoms associated with disease caused by Corynebacterium diphtheriae (diphtheria), Bordetella pertussis (whooping cough), Vibrio cholerae (cholera), Bacillus anthracis (anthrax), Clostridium botulinum (botulism), Clostridium tetani (tetanus), and enterohemorrhagic Escherichia coli (bloody diarrhea and hemolytic uremic syndrome) are all related to the activities of the toxins produced by these organisms. With the recognition of the central role of toxin in these and other diseases has come the application of inactive toxins (toxoids) as vaccines. Such toxoid vaccines have had an important positive impact on public health.
In this review, we provide a summary overview (Table) of a variety of bacterial toxins categorized according to mode of action: damaging cell membranes, inhibiting protein synthesis, activating second messenger pathways, inhibiting the release of neurotransmitters, or activating the host immune response. We also describe in detail seven toxins: Staphylococcus aureus α-toxin, Shiga toxin (Stx), cytotoxic necrotizing factor type 1 (CNF1), E. coli heat-stable toxin (ST), botulinum and tetanus neurotoxins, and toxic-shock syndrome toxin (TSST) produced by S. aureus. We emphasize these toxins because they are produced by emerging (Stx of enterohemorrhagic E. coli) or reemerging (α-toxin of multidrug-resistant S. aureus) pathogens or illustrate different structures or modes of action (ST, CNF1, neurotoxins, and TSST).
| Organism/toxin | Mode of action | Target | Disease | Toxin implicated in diseaseb |
|---|---|---|---|---|
| Damage membranes | ||||
| Aeromonas hydrophila/aerolysin | Pore-former | Glycophorin | Diarrhea | (yes) |
| Clostridium perfringens/ | Pore-former | Cholesterol | Gas gangrenec | ? |
| perfringolysin O | ||||
| Escherichia coli/ hemolysind | Pore-former | Plasma membrane | UTIs | (yes) |
| Listeria monocytogenes/ | Pore-former | Cholesterol | Foodborne systemic | (yes) |
| listeriolysin O | illness meningitis | |||
| Staphyloccocus aureus/ α-toxin | Pore-former | Plasma membrane | Abcessesc | (yes) |
| Streptococcus pneumoniae/ | Pore-former | Cholesterol | Pneumoniac | (yes) |
| pneumolysin | ||||
| Streptococcus pyogenes/ | Pore-former | Cholesterol | Strep throat Sfc | ? |
| streptolysin O | ||||
| Inhibit protein synthesis | ||||
| Corynebacterium diphtheriae/ | ADP-ribosyltransferase | Elongation factor 2 | Diphtheria | yes |
| diphtheria toxin | ||||
| E. coli/Shigella dysenteriae/ | N-glycosidase | 28S rRNA | HC and HUS | yes |
| Shiga toxins | ||||
| Pseudomonas aeruginosa/ | ADP-ribosyltransferase | Elongation factor 2 | Pneumoniac | (yes) |
| exotoxin A | ||||
| Activate second messenger pathways | ||||
| E.coli | ||||
| CNF | Deamidase | Rho G-proteins | UTIs | ? |
| LT | ADP-ribosyltransferase | G-proteins | Diarrhea | yes |
| STd | Stimulates guanylate cyclase | guanylate cyclase receptor | Diarrhea | yes |
| CLDTd | G2 block | Unknown | Diarrhea | (yes) |
| EAST | ST-like? | Unknown | Diarrhea | ? |
| Bacillus anthracis/edema factor | Adenylate cyclase | ATP | Anthrax | yes |
| Bordetella pertussis/ | ||||
| dermonecrotic toxin | Deamidase | Rho G-proteins | Rhinitis | (yes) |
| pertussis toxin | ADP-ribosyltransferase | G-protein(s) | Pertussis | yes |
| Clostridium botulinum/ C2 toxin | ADP-ribosyltransferase | Monomeric G-actin | Botulism | ? |
| C. botulinum/C3 toxin | ADP-ribosyltransferase | Rho G-protein | Botulism | ? |
| Clostridium difficile/ | ||||
| toxin A | Glucosyltransferase | Rho G-protein(s) | Diarrhea/PC | (yes) |
| toxin B | Glucosyltransferase | Rho G-protein(s) | Diarrhea/PC | ? |
| Vibrio cholerae/cholera toxin | ADP-ribosyltransferase | G-protein(s) | Cholera | yes |
| Activate immune response | ||||
| S. aureus/ | ||||
| enterotoxins | Superantigen | TCR and MHC II | Food poisoningc | yes |
| exfoliative toxins | Superantigen (and serine protease?) | TCR and MHC II | SSSc | yes |
| toxic-shock toxin | Superantigen | TCR and MHC II | TSSc | yes |
| S. pyogenes/pyrogenic exotoxins | Superantigens | TCR and MHC II | SF/TSSc | yes |
| Protease | ||||
| B. anthracis/lethal factor | Metalloprotease | MAPKK1/MAPKK2 | Anthrax | yes |
| C. botulinum/neurotoxins A-G | Zinc-metalloprotease | VAMP/synaptobrevin =SNAP-25 syntaxin | Botulism | yes |
| Clostridium tetani/ tetanus toxin | Zinc-metalloprotease | VAMP/synaptobrevin | Tetanus | yes |
aAbbreviations: CNF, cytotoxic necrotizing factor; LT, heat-labile toxin; ST, heat-stable toxin; CLDT, cytolethal distending toxin; EAST, enteroaggregative
Many bacterial exotoxins have the capacity to damage the extracellular matrix or the plasma membrane of eukaryotic cells. The damage not only may result in the direct lysis of cells but also can facilitate bacterial spread through tissues. Toxins that mediate this cellular damage do so by either enzymatic hydrolysis or pore formation. Bacterial hyaluronidases, collagenases, and phospholipases have the capacity to degrade cellular membranes or matrices. Specific examples of these types of toxins include the α-toxin of Clostridium perfringens, which has phospholipase C activity; Streptococcus pyogenes streptokinase, which can hydrolyze plasminogen to plasmin and dissolve clots; and the clostridial collagenases (3-5). Pore-forming toxins, as the name suggests, disrupt the selective influx and efflux of ions across the plasma membrane by inserting a transmembrane pore. This group of toxins includes the RTX (repeats in toxin) toxins from gram-negative bacteria, streptolysin O produced by S. pyogenes, and the S. aureus α-toxin (described below).
Figure 1
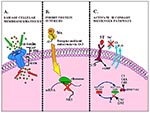
Figure 1. Diagrammatic representation of the mode of action of several bacterial toxins. A. Damage to cellular membranes by Staphylococcus aureus α-toxin. After binding and oligomerization, the stem of the mushroom-shaped α-toxin heptamer...
S. aureus α-toxin can be considered the prototype of oligomerizing pore-forming cytotoxins. The α-toxin gene resides as a single copy on the chromosome of most pathogenic S. aureus strains, and its expression is environmentally regulated at the transcriptional level by the staphylococcal accessory gene regulator (agr) locus (6,7). The α-toxin is synthesized as a 319 amino acid precursor molecule that contains an N-terminal signal sequence of 26 amino acids. The secreted mature toxin, or protomer, is a hydrophilic molecule that lacks cysteine residues and has a molecular mass of approximately 33 kDa (6-8). Recently, the crystallographic structure of the fully assembled α-toxin pore was solved (9). On the plasma membrane, seven toxin protomers assemble to form a 232 kDa mushroom-shaped heptamer comprising three distinct domains (Figure 1A) (9,10). The cap and rim domains of the α-toxin heptamer are situated at the surface of the plasma membrane, while the stem domain serves as the transmembrane channel.
Alpha-toxin is cytolytic to a variety of cell types, including human monocytes, lymphocytes, erythrocytes, platelets, and endothelial cells (6,8). For α-toxin to damage cellular membranes, three sequential events are required. Toxin protomers must first bind to target membranes by either unidentified high-affinity receptors or through nonspecific absorption to substances such as phosphotidylcholine or cholesterol on the lipid bilayer (6-8). Second, membrane-bound protomers must oligomerize into a nonlytic prepore heptamer complex. Third, the heptamer must undergo a series of conformational changes that create the stem domain of the toxin, which is then inserted into the membrane (9,10). The α-toxin pore allows the influx and efflux of small molecules and ions that eventually lead to the swelling and death of nucleated cells and the osmotic lysis of erythrocytes. Pore formation has also been shown to trigger secondary events that could promote development of pathologic sequelae. These events include endonuclease activation, increased platelet exocytosis, release of cytokines and inflammatory mediators, and production of eicosanoids (6,8). Several animal models have demonstrated that α-toxin is required for S. aureus virulence in these systems (6,8); however, the precise role of α-toxin in staphylococcal diseases in humans remains unclear.
A second class of toxins intoxicates target cells by inhibiting protein synthesis. Substrates for toxins in this group are elongation factors and ribosomal RNA. Diphtheria toxin and Pseudomonas exotoxin A act by ADP-ribosylating elongation factor 2 (EF2) (11,12). The modified EF2 is no longer able to function in protein synthesis. Stxs, also called verotoxins, are produced by Shigella dysenteriae serotype 1 and the emerging pathogens designated Stx-producing E. coli (STEC). Stxs inactivate ribosomal RNA (by a mechanism described below) so that the affected ribosome can no longer interact with elongation factors (13,14). The inhibition of protein synthesis by this group of toxins ultimately results in death of the target cell.
Stxs are potent cytotoxins that can be divided into two antigenically distinct groups that share 50% to 60% homology: Stx/Stx1 and Stx2 (15-17). Stx and Stx1 are elaborated by S. dysenteriae serotype 1 and E. coli, respectively, and differ at only one amino acid. Stx2-type toxins have been found only in E. coli isolates and are quite diverse. While Stx2 is considered the prototype of this group, variants have been found that differ antigenically, in receptor specificity and in activation by intestinal mucus. Some of these attributes are the result of only one or two nucleotide differences in the toxin genes.
The stx of S. dysenteriae is invariably chromosomally located. The genes that encode Stx1 and Stx2 are carried chromosomally or by lysogenic bacteriophages. The genes that code for the A and B subunits of Stxs, stxA and stxB, are organized within an operon. The operator region of Stx/Stx1 (but not Stx2) contains a consensus fur box that is responsible for the iron-regulation of Stx and Stx1 production. Neither iron nor any other environmental factors examined affect the expression of Stx2. However, intestinal mucus enhances the activity of some Stx2 variants (18). The Stxs, which carry typical N-terminal leader sequences, are not actively secreted from the bacterial cell and are thought to be released into the milieu during cell lysis.
Figure 2

Stxs display an AB-toxin structure; an enzymatically active A subunit is noncovalently associated with a binding, or B, component. The crystal structures of the Stx1 B pentamer (19) and the Stx holotoxin have been solved (20) (Figure 2). Other toxins that share this AB structure are the E. coli heat-labile toxin (21), cholera toxin, and pertussis toxin (22) (Figure 2). The molecular masses of mature Stx A and B monomeric subunits are approximately 35 kDa and 7.5 kDa, respectively, although holotoxin contains five B subunit molecules. The B subunit pentamer directs the binding of the holotoxin to sensitive eukaryotic cells via specific glycolipid receptors. Once internalized, the A polypeptide is cleaved into an enzymatically active A1 portion and an A2 portion; these fragments remain associated through a disulfide bond. The A2 portion serves to link the A1 fragment and the B pentamer.
The enzymatic A subunit acts as a specific N-glycosidase to cleave a single adenine residue from 28S ribosomal RNA (13,14). This depurination ultimately results in the inhibition of protein synthesis within intoxicated cells (Figure 1B). Prokaryotic ribosomes are as sensitive to the N-glycosidase activity of Stx as eukaryotic ribosomes (23).
STEC are considered emerging pathogens (24) because they were first described less than 20 years ago, during a 1983 outbreak of hemorrhagic colitis associated with undercooked hamburger (25,26). STEC O157:H7 causes approximately 20,000 cases of hemorrhagic colitis each year in the United States (27). Approximately 1,000 cases of the life-threatening sequelae hemolytic uremic syndrome and approximately 100 deaths are also attributed to E. coli O157:H7 annually in the United States (27).
Bacterial toxins can also target and alter the function of a variety of cellular proteins without directly killing the intoxicated cell. Toxin activation or modification of secondary messengers can cause dramatic alterations to signal transduction pathways critical in maintaining a variety of cellular functions. To demonstrate the diversity among the toxins that belong to this category, we will describe CNF type 1 and the heat-stable enterotoxins.
Cytotoxic Necrotizing Factor (CNF)
CNF types 1 and 2 (CNF1/2) from E. coli belong to a group of bacterial toxins that modify Rho, a subfamily of small GTP-binding proteins that are regulators of the actin cytoskeleton (28,29). Most members of this toxin family, which includes the large clostridial cytotoxins and the C3 exoenzyme of C. botulinum, inactivate Rho (29). CNF1, CNF2, and the dermonecrotic toxins from Bordetella species form a unique subset in this family, since these toxins have the capacity to activate Rho (Figure 1C) (29-32). CNF1 and CNF2 share 99% amino acid similarity; however, we will discuss only CNF1 in detail because of its association with extraintestinal E. coli infections in humans, most notably urinary tract infections.
The gene for CNF1 is chromosomally encoded and resides on a pathogenicity island in uropathogenic E. coli (33,34). The toxin is synthesized as a hydrophilic polypeptide of approximately 115 kDa that remains primarily cytoplasmic because of the lack of a signal sequence (33). Recent structure and function analysis of CNF1 indicates that the toxin has distinct binding and enyzmatic domains (35). The N-terminal half of CNF1, which includes two potential transmembrane domains, contains the cellular binding domain. This region of the molecule shows amino acid similarity to the Pasteurella multicoda toxin, a potent mitogen thought to be the etiologic agent of progressive atrophic rhinitis in pigs (33,35). The C-terminal portion of CNF1 represents the toxin's enzymatic domain and shows homology with dermonecrotic toxins in a 100-amino acid stretch that may represent the active site of the toxin (33,35).
Figure 3

Figure 3. The effect of cytotoxic necrotizing factor type 1 (CNF1) on eukaryotic cells. A. HEp-2 cells, magnification 10X. B. HEp-2 cells intoxicated with CNF1, magnification 10X.
Eukaryotic cells intoxicated with CNF1 exhibit membrane ruffling; the formation of focal adhesions and actin stress fibers; and DNA replication in the absence of cell division, a phenomenon that results in enlarged multinucleated cells (Figure 3). The drastic changes apparent in CNF1-treated cells are a result of the toxin's capacity to modify Rho (29,30,32). This modification has recently been identified as a deamidation of the glutamine residue at position 63 of Rho to a glutamic acid. This amino acid change produces a dominant active Rho protein unable to hydrolyze bound GTP (30,32). In vivo, CNF1 causes necrosis in rabbit skin following intradermal injection and persistent inflammation in a mouse footpad assay (36). Epidemiologic data support the role of CNF1 as a virulence factor in human extraintestinal infections, although direct proof of the toxin's role in disease remains to be determined (29,37).
Heat-Stable Toxin (ST)
Two families of diarrheagenic STs have been described: STa (or STI) and STb (or STII). Distinct STas are produced by a variety of enteric pathogenic organisms: enterotoxinogenic E. coli (ETEC) (the focus of this section), V. cholerae, Vibrio mimicus, Yersinia enterocolitica, Citrobacter freundii, and Klebsiella.
Strains of ETEC associated with human disease may produce either STa, heat-labile toxin I, or both. STas from ETEC isolates are related but distinct toxins (38). STh is produced by strains of human origin, while STp is found predominantly in porcine strains. The STa genes (estA) of ETEC are encoded within a transposable element and have been found on a variety of replicons (39,40). STa is translated as a precursor molecule of 72 amino acids and undergoes two cleavage events before the secretion of the mature form into the culture supernatant. Mature STs are small peptides that range from 17 to 53 amino acids. STh and STp contain 19 and 18 residues, respectively. STas share a conserved C-terminal region of 13 amino acids essential for toxicity and the heat-stable nature of the toxin. Six cysteine residues are present within this domain, and the three disulphide bonds formed between the cysteine residues are necessary for toxicity of the molecule. Binding of STa to its cellular receptor results in the stimulation of membrane-bound guanylate cyclase, which in turn leads to an increase in intracellular cyclic GMP (Figure 1C) (41). This increase in cyclic GMP affects electrolyte flux in the bowel; sodium absorption is inhibited and chloride secretion is stimulated. These ion flux changes result in the secretory diarrhea characteristic of ETEC infection. ETEC cause traveler's diarrhea and are a major source of childhood diarrhea in many parts of the world.
The C. botulinum neurotoxins (BoNTs, serotypes A-G) and the C. tetani tetanus neurotoxin (TeNT) constitute another category of bacterial toxins on the basis of similarities in structure, enzymatic activity, and the targeting to cells of the nervous system. BoNTs are most commonly associated with infant and foodborne botulism and exist in nature as large complexes comprised of the neurotoxin and one or more associated proteins believed to provide protection and stability to the toxin molecule while in the gut (42,43). TeNT, which is synthesized from vegetative C. tetani in wounds, does not appear to form complexes with any other protein components (42,43).
The BoNTs and TeNT are either plasmid encoded (TeNT, BoNTs/A, G, and possibly B) or bacteriophage encoded (BoNTs/C, D, E, F), and the neurotoxins are synthesized as inactive polypeptides of 150 kDa (44). BoNTs and TeNT are released from lysed bacterial cells and then activated by the proteolytic cleavage of an exposed loop in the neurotoxin polypeptide (45). Each active neurotoxin molecule consists of a heavy (100 kDa) and light chain (50 kDa) linked by a single interchain disulphide bond (42,45). The heavy chains of both the BoNTs and TeNT contain two domains: a region necessary for toxin translocation located in the N-terminal half of the molecule, and a cell-binding domain located within the C-terminus of the heavy chain (45,46). The light chains of both the BoNTs and TeNT contain zinc-binding motifs required for the zinc-dependent protease activities of the molecules (45,46).
The cellular targets of the BoNTs and TeNT are a group of proteins required for docking and fusion of synaptic vesicles to presynaptic plasma membranes and therefore essential for the release of neurotransmitters. The BoNTs bind to receptors on the presynaptic membrane of motor neurons associated with the peripheral nervous system. Proteolysis of target proteins in these neurons inhibits the release of acetylcholine, thereby preventing muscle contraction (47,48). BoNTs/B, D, F, and G cleave the vesicle-associated membrane protein and synaptobrevin, BoNT/A and E target the synaptosomal-associated protein SNAP-25, and BoNT/C hydrolyzes syntaxin and SNAP-25 (42,45,46). TeNT affects the central nervous system and does so by entering two types of neurons. TeNT initially binds to receptors on the presynaptic membrane of motor neurons but then migrates by retrograde vesicular transport to the spinal cord, where the neurotoxin can enter inhibitory interneurons (45,47). Cleavage of the vesicle-associated membrane protein and synaptobrevin in these neurons disrupts the release of glycine and gamma-amino-butyric acid, which, in turn, induces muscle contraction (47,48). The contrasting clinical manifestations of BoNT or TeNT intoxication (flaccid and spastic paralysis, respectively) are the direct result of the specific neurons affected and the type of neurotransmitters blocked (45-47).
Several bacterial toxins can act directly on the T cells and antigen-presenting cells of the immune system. Impairment of the immunologic functions of these cells by toxin can lead to human disease. One large family of toxins in this category are the pyrogenic toxin superantigens (PTSAgs), whose hallmark biological activities include potent stimulation of the immune cell system, pyrogenicity, and enhancement of endotoxin shock (49-51). These stable, secreted toxins of 22 kDa to 30 kDa include staphylococcal enterotoxins serotypes A-E, G, and H; group A streptococcal pyrogenic exotoxins serotypes A-C and F; group A streptococcal superantigen; and staphylococcal TSST-1, which we discuss below.
All PTSAgs share common biological activities, but TSST-1 is the most divergent member of the toxin family, with less than 30% amino acid homology to other family members (52-54). TSST-1 is chromosomally encoded, and the tst gene is located in a variable genetic element in S. aureus (49,52,55). The toxin is synthesized as a precursor molecule of 234 residues with the first 40 amino acids acting as a signal sequence that is cleaved to generate the mature 22 kDa toxin (49). Expression of TSST-1 depends on oxygen, temperature, pH and glucose levels, and is regulated by the S. aureus agr locus (49,51). On the basis of crystallographic analysis, TSST-1 appears structurally similar to several other PTSAgs in that the toxin consists of two distinct domains; however, unlike other family members, TSST-1 does not require a zinc cofactor (51-54). Domain A of TSST-1 (amino acid residues 1-17 and 90-194) exists as a ß-grasp motif, and domain B consists of a five-stranded ß-barrel motif that forms an oligosaccharide/oligonucleotide binding fold.
In general, the potent immunostimulatory properties of PTSAgs are a direct result of toxin binding to distinct regions outside the peptide binding cleft of the major histocompatibility class II molecules (expressed on the surface of antigen-presenting cells) and to specific Vß elements on the T-cell receptor. In particular, the domain B of TSST-1 binds primarily to the a-chain of human leukocyte antigen-DR1 molecules, while domain A specifically binds to human T-cell receptor Vß2 elements (51-53,56). Binding of TSST-1 to Vß2 T-cell receptor elements results in a massive proliferation of up to 20% of peripheral T cells, an event that drastically skews the T-cell Vß repertoire (53,56). T cells that undergo this expansion can subsequently exist in a state of anergy or undergo apoptosis (56). Concomitant to T-cell proliferation is a massive release of both lymphocyte (interleukin [IL]-2, tumor necrosis factor ß, gamma interferon)-derived and monocyte (IL-1, IL-6, tumor necrosis factor a)-derived cytokines (51,56). These cytokines serve as mediators of the hypotension, high fever, and diffuse erythematous rash that are characteristic of toxic-shock syndrome. Long established as a key substance in causing staphylococcal toxic-shock syndrome, TSST-1 has more recently been linked with Kawasaki syndrome, a leading cause of acquired heart disease in children in the United States (50,54).
Some of these powerful disease-causing toxins have been exploited to further basic knowledge of cell biology or for medical purposes. For example, cholera toxin and the related labile-toxin of E. coli, as well as B. pertussis toxin, have been used as biologic tools to understand the mechanism of adenylate cyclase activation and the role of cyclic AMP as a second messenger in the eukaryotic cell (57-59). Derivatives of some of these toxins, cholera toxin and labile toxin, have also been incorporated into human vaccines because of the adjuvant properties of these molecules (60,61).
Similarly, the activities of several potent cytotoxins have been harnessed as potential therapies for certain cancers. Such toxins can either be used directly in treatment or as components of immunotoxins (62-64). For example, Stx binds to the cell surface glycolipid CD77, which is expressed by B cells in certain B-cell lymphomas (65,66). This finding led to studies that showed that Stx can purge murine (and potentially human) bone marrow of malignant CD77+ B cells before an autologous bone marrow transplant (67). Other toxins that inhibit protein synthesis, such as diphtheria toxin, Pseudomonas exotoxin A, or the plant toxin ricin, are frequently engineered as the cell-killing component of immunotoxins. These "magic bullets," hybrids of the enzymatically active portion of a toxin molecule and monoclonal antibodies (or a receptor), are in clinical trials for the treatment of persons with B-cell lymphomas, leukemia, and bone marrow transplants.
Several clinical applications have also been found for the powerful botulinum neurotoxin type A (BoNT/A) (46,68). The disorders that respond to BoNT/A involve muscle hyperactivity. A minuscule amount of purified toxin injected into specific sites results in paralysis of the target muscle and ablation of the muscle spasm. Therapy must be continual since the effect of the toxin usually lasts for no more than several months. The first maladies treated with BoNT/A were eye movement abnormalities (69). However, the therapeutic value of BoNT/A has been shown for many other disorders including cervical and laryngeal dystonia, writer's cramp, hemifacial spasm, tremors, and tics (46,68). BoNT/A is also used cosmetically to reduce deep wrinkles caused by the contraction of facial muscles (70).
Another toxic bacterial product with medical applications is streptokinase, a potent plasminogen activator produced by several pathogenic streptococcal strains. The proteolytic activity of streptokinase is used to clear blocked arteries in patients who have heart attacks (71,72).
Vaccines directed at the toxic component of bacterial pathogens have proven quite effective in preventing certain diseases. Most licensed toxoid vaccines are relatively crude, but effective, preparations. These vaccines consist of partially purified toxin preparations obtained from culture supernatants of bacteria such as C. diphtheriae, C. tetani, or B. anthracis. Formaldehyde treatment is used to detoxify the diphtheria and tetanus toxins for vaccine formulation. The anthrax vaccine contains the protective antigen and small amounts of the lethal factor and edema factor toxins. The current botulinum vaccine is an investigational drug composed of crude preparations of five botulinum toxoids and is distributed by the Centers for Disease Control and Prevention to researchers that work with the toxin or organism. Acellular pertussis vaccines that contain pertussis toxoid, alone or as one of several components, are as effective as killed whole-cell vaccines but less reactogenic (73); such vaccines have recently been approved for use in infants as well as older children.
New vaccines aimed at toxins are in various stages of development: research and development, preclinical, phase I, phase II, or phase III (74). The next generation of toxoid vaccines falls into three general categories: purified toxoids that have been inactivated by chemical or genetic means; live, attenuated strains of the causative agent that produce a genetically derived toxoid; or live, attenuated unrelated bacterial vector strains, such as V. cholerae or Salmonella, that produce the target toxoid. Examples of each of these approaches and progress in development of specific toxoid vaccines are described annually in the Jordan Report (74).
Antitoxins raised against diphtheria, tetanus, and botulinum toxoids have also been used for many years to treat seriously ill patients. Antiserum specific for the Stx toxins produced by E. coli O157:H7 and other STEC is under development for the treatment and prevention of hemolytic uremic syndrome, a life-threatening sequela of these infections.
Microbial toxins capable of interrupting or hyperstimulating many essential functions and pathways of eukaryotic cells have evolved along with the carrier bacterium. Presumably these toxins confer some benefit to the bacterium, either during a stage of the host-parasite interaction or in some environmental niche encountered by the bacterium. Certain bacterial toxins act on the target cell surface to irreparably damage the cell membrane or alter normal cellular signal transduction. Other toxins exhibit enzymatic activity once the molecule has gained access to the cytoplasm of the sensitive cell by endocytosis. Yet other bacterial toxins act by either turning off or locking on a normal host cell function.
Although detrimental to the susceptible host during an infection, the activities of several bacterial toxins have been exploited as probes of eukaryotic cellular pathways and for medicinal applications. Thus, research on a microbial toxin produced by an established, emerging, or reemerging pathogen is likely to yield novel information about the role of that toxin in disease as well as the properties of host cells that are subverted by the toxin.
Dr. Schmitt is a research assistant professor in the Department of Microbiology and Immunology, Uniformed Services University of the Health Sciences, Bethesda, Maryland, USA. Research interests include virulence factors and pathogenesis of disease caused by Shiga toxin-producing E. coli and regulation of Salmonella virulence.
Acknowledgments
We thank Drs. Ethan Merritt and Marie Frasier for generating and contributing the figures on toxin crystal structures.
Work in Dr. O'Brien's laboratory is supported by grants from the National Institutes of Health (AI20148-16, AI33525-5, AI38281-3), the Department of Agriculture (97-35201-4578), and the Uniformed Services University of the Health Sciences (R073EQ).
References
- Roux E, Yersin A. Contribution a l'etude de la diphtherie. Ann Inst Pasteur (Paris). 1888;2:629–61.
- Schlessinger D, Schaechter M. Bacterial toxins. In: Schaechter M, Medoff G, Eisenstein BI, editors. Mechanisms of microbial disease. 2nd ed. Baltimore: Williams and Wilkins; 1993. p. 162-75.
- Songer JG. Bacterial phospholipases and their role in virulence. Trends Microbiol. 1997;5:156–61. DOIPubMedGoogle Scholar
- Lottenberg R, Minning-Wenz D, Boyle MD. Capturing host plasmin(ogen): a common mechanism for invasive pathogens? Trends Microbiol. 1994;2:20–4. DOIPubMedGoogle Scholar
- Harrington DJ. Bacterial collagenases and collagen-degrading enzymes and their potential role in human disease. Infect Immun. 1996;64:1885–91.PubMedGoogle Scholar
- Bhakdi S, Tranum-Jensen J. Alpha-toxin of Staphylococcus aureus. Microbiol Rev. 1991;55:733–51.PubMedGoogle Scholar
- Tomita T, Kamio Y. Molecular biology of the pore-forming cytolysins from Staphylococcus aureus, a- and gamma-hemolysins and leukocidin. Biosci Biotechnol Biochem. 1997;61:565–72. DOIPubMedGoogle Scholar
- Bhakdi S, Bayley H, Valeva A, Walev I, Walker B, Weller U, Staphylococcal alpha-toxin, streptolysin-O and Escherichia coli hemolysin: prototypes of pore-forming bacterial cytolysins. Arch Microbiol. 1996;165:73–9. DOIPubMedGoogle Scholar
- Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–66. DOIPubMedGoogle Scholar
- Lesieur C, Vecsey-Semjen B, Abrami L, Fivaz M, Gisou van der Goot F. Membrane insertion: the strategies of toxins. Mol Membr Biol. 1997;14:45–64. DOIPubMedGoogle Scholar
- Collier RJ. In: Moss J, Vaughan M, editors. ADP-ribosylating toxins and g proteins. Washington: American Society for Microbiology; 1990. p. 3-19.
- Wick MJ, Iglewski BH. In: Moss J, Vaughan M, editors. ADP-ribosylating toxins and g proteins. Washington: American Society for Microbiology; 1990. p. 31-43.
- Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara Y, Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eucaryotic ribosomes. Eur J Biochem. 1988;171:45–50. DOIPubMedGoogle Scholar
- Saxena SK, O'Brien AD, Ackerman EJ. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28 S RNA when microinjected into Xenopus oocytes. J Biol Chem. 1989;264:596–601.PubMedGoogle Scholar
- Tesh VL, O'Brien AD. The pathogenic mechanisms of Shiga toxin and the Shiga-like toxins. Mol Microbiol. 1991;5:1817–22. DOIPubMedGoogle Scholar
- O'Brien AD, Tesh VL, Donohue-Rolfe A, Jackson MP, Olsnes S, Sandvig K, Shiga toxin: biochemistry, genetics, mode of action, and role in pathogenesis. In: Sansonetti PJ, editor. Pathogenesis of shigellosis. 180th ed. Berlin-Heidelberg: Springer-Verlag; 1992. p. 66-94.
- O'Brien AD, Kaper JB. Shiga toxin-producing Escherichia coli: yesterday, today, and tomorrow. In: Kaper JB, O'Brien AD, editors. Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. Washington: American Society for Microbiology; 1998. p. 1-11.
- Melton-Celsa AR, O'Brien AD. Activation of Shiga-like toxins by mouse and human intestinal mucus correlates with virulence of enterohemorrhagic Escherichia coli O91:H21 isolates in orally infected, streptomycin-treated mice. Infect Immun. 1996;64:1569–76.PubMedGoogle Scholar
- Stein PE, Boodhoo A, Tyrell GT, Brunton J, Read RJ. Crystal structure of the cell-binding B oligomer of verotoxin-1 from E. coli. Nature. 1992;355:748–50. DOIPubMedGoogle Scholar
- Frasier ME, Chernaia MM, Kozlov YV, James MNG. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat Struct Biol. 1994;1:59–64. DOIPubMedGoogle Scholar
- Sixma TK, Kalk KH, van Zanten BA, Dauter Z, Kingma J, Witholt B, Redefined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J Mol Biol. 1993;230:890–918. DOIPubMedGoogle Scholar
- Stein PE, Boodhoo A, Armstrong GD, Cockle SA, Klein MH, Read RJ. The crystal structure of pertussis toxin. Structure. 1994;2:45–57. DOIPubMedGoogle Scholar
- Suh J-K, Hovde CJ, Robertus JD. Shiga toxin attacks bacterial ribosomes as effectively as eukaryotic ribosomes. Biochemistry. 1998;37:9394–8. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Addressing emerging infectious disease threats: a prevention strategy for the United States. MMWR Morb Mortal Wkly Rep. 1994;43:1–18.
- O'Brien AD, Lively TA, Chen M, Rothman SW, Formal SB. Escherichia coli O157:H7 strains associated with hemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (Shiga) like cytotoxin. Lancet. 1983;i:702. DOIGoogle Scholar
- Centers for Disease Control. Isolation of E. coli O157:H7 from sporadic cases of hemorrhagic colitisUnited States. MMWR Morb Mortal Wkly Rep. 1982;31:580–5.PubMedGoogle Scholar
- Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333:364–8. DOIPubMedGoogle Scholar
- Aktories K. Rho proteins: targets for bacterial toxins. Trends Microbiol. 1997;5:282–8. DOIPubMedGoogle Scholar
- Oswald E, Sugai M, Labigne A, Wu HC, Fiorentini C, Boquet P, Cytotoxic necrotizing factor type 2 produced by virulent Escherichia coli modifies the small GTP-binding proteins Rho involved in assembly of actin stress fibers. Proc Natl Acad Sci U S A. 1994;91:3814–8. DOIPubMedGoogle Scholar
- Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387:725–9. DOIPubMedGoogle Scholar
- Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature. 1997;387:729–33. DOIPubMedGoogle Scholar
- Horiguchi Y, Inoue N, Masuda M, Kashimoto T, Katahira J, Sugimoto N, Bordetella bronchiseptica dermonecrotizing toxin induces reorganization of actin stress fibers through deamidation of Gln-63 of the GTP-binding protein Rho. Proc Natl Acad Sci U S A. 1997;94:11623–6. DOIPubMedGoogle Scholar
- Falbo V, Pace T, Picci L, Pizzi E, Caprioli A. Isolation and nucleotide sequence of the gene encoding cytotoxic necrotizing factor 1 of Escherichia coli. Infect Immun. 1993;61:4909–14.PubMedGoogle Scholar
- Blum G, Falbo V, Caprioli A, Hacker J. Gene clusters encoding the cytotoxic necrotizing factor type 1, Prs-fimbriae and -hemolysin form the pathogenicity island II of the uropathogenic Escherichia coli strain J96. FEMS Microbiol Lett. 1995;126:189–96.PubMedGoogle Scholar
- Lemichez E, Flatau G, Bruzzone M, Boquet P, Gauthier M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol Microbiol. 1997;24:1061–70. DOIPubMedGoogle Scholar
- DeRycke J, Gonzalez EA, Blanco J, Oswald E, Blanco M, Boivin R. Evidence for two types of cytotoxic necrotizing factor in human and animal clinical isolates of Escherichia coli. J Clin Microbiol. 1990;28:694–9.PubMedGoogle Scholar
- Andreu A, Stapleton AE, Fennell C, Lockman HA, Xercavins M, Fernandez F, Urovirulence determinants in Escherichia coli strains causing prostatitis. J Infect Dis. 1997;176:464–9. DOIPubMedGoogle Scholar
- Nair GB, Takeda Y. The heat-stable enterotoxins. Microb Pathog. 1998;24:123–31. DOIPubMedGoogle Scholar
- So M, McCarthy BJ. Nucleotide sequence of transposon Tn1681 encoding a heat-stable toxin (ST) and its identification in enterotoxigenic Escherichia coli strains. Proc Natl Acad Sci U S A. 1980;77:4011–5. DOIPubMedGoogle Scholar
- So M, Boyer HW, Betlach M, Falkow S. Molecular cloning of an Escherichia coli plasmid determinant that encodes for the production of heat-stable enterotoxin. J Bacteriol. 1976;128:463–72.PubMedGoogle Scholar
- Giannella RA. Escherichia coli heat-stable enterotoxins, guanylins, and their receptors: what are they and what do they do? J Lab Clin Med. 1995;125:173–81.PubMedGoogle Scholar
- Singh BR, Li B, Read D. Botulinum versus tetanus neurotoxins: why is botulinum neurotoxin but not tetanus neurotoxin a food poison? Toxicon. 1995;33:1541–7. DOIPubMedGoogle Scholar
- Jahn R, Hanson PI, Otto H, Ahnert-Hilger G. Botulinum and tetanus neurotoxins: emerging tools for the study of membrane fusion. Cold Spring Harb Symp Quant Biol. 1995;60:329–35.PubMedGoogle Scholar
- Henderson I, Davis T, Elmore M, Minton NP. The genetic basis of toxin production in Clostridium botulinum and Clostridium tetani. In: Rood JI, McClane BA, Songer JG, Titball RW, editors. The clostridia: molecular biology and pathogenesis. San Diego: Academic Press; 1997. p. 261-94.
- Schiavo G, Montecucco C. The structure and mode of action of botulinum and tetanus toxins. In: Rood JI, McClane BA, Songer JG, Titball RW, editors. The clostridia: molecular biology and pathogenesis. San Diego: Academic Press; 1997. p. 295-322.
- Kessler KR, Benecke R. Botulinum toxin: from poison to remedy. Neurotoxicology. 1997;18:761–70.PubMedGoogle Scholar
- Halpern JL, Neale EA. Neurospecific binding, internalization and retrograde axonal transport. Curr Top Microbiol Immunol. 1995;195:221–41.PubMedGoogle Scholar
- Arnon SS. Human tetanus and human botulism. In: Rood JI, McClane BA, Songer JG, Titball RW, editors. The clostridia: molecular biology and pathogenesis. San Diego: Academic Press; 1997. p. 95-115.
- Rago JV, Schlievert PM. Mechanisms of pathogenesis of staphylococcal and streptococcal superantigens. Curr Top Microbiol Immunol. 1998;225:81–97.PubMedGoogle Scholar
- Lee PK, Schlievert PM. Molecular genetics of pyrogenic exotoxin "superantigens" of Group A streptococci and staphylococcus. Curr Top Microbiol Immunol. 1991;174:1–19.PubMedGoogle Scholar
- Bohach GA, Stauffacher CV, Ohlendorf DH, Chi YI, Vath GM, Schlievert PM. The staphylococcal and streptococcal pyrogenic toxin family. In: Singh BR, Tu AT, editors. Natural Toxins II. New York: Plenum Press; 1996. p. 131-54.
- Papageorgiou AC, Acharya KR. Superantigens as immunomodulators: recent structural insights. Structure. 1997;5:991–6. DOIPubMedGoogle Scholar
- Prasad GS, Radhakrishnan R, Mitchell DT, Earhart CA, Dinges MM, Cook WJ, Refined structures of three crystal forms of toxic shock syndrome toxin-1 and of a tetramutant with reduced activity. Protein Sci. 1997;6:1220–7. DOIPubMedGoogle Scholar
- Betley MJ, Borst DW, Regassa LB. Staphylococcal enterotoxins, toxic shock syndrome toxin and streptococcal pyrogenic exotoxins: a comparative study of their molecular biology. Chem Immunol. 1992;55:1–35.PubMedGoogle Scholar
- Stevens DL. Superantigens: their role in infectious diseases. Immunol Invest. 1997;26:275–81. DOIPubMedGoogle Scholar
- Harnett MM. Analysis of G-proteins regulating signal transduction pathways. Methods Mol Biol. 1994;27:199–211.PubMedGoogle Scholar
- Bokoch GM, Katada T, Northup JK, Hewlett EL, Gilman AG. Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J Biol Chem. 1983;258:2072–5.PubMedGoogle Scholar
- Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80:249–57. DOIPubMedGoogle Scholar
- Snider DP. The mucosal adjuvant activities of ADP-ribosylating bacterial enterotoxins. Crit Rev Immunol. 1995;15:317–48.PubMedGoogle Scholar
- Holmgren J, Lycke N, Czerkinsky C. Cholera toxin and cholera-B subunit as oral mucosal adjuvant and antigen vector systems. Vaccine. 1993;11:1179–84. DOIPubMedGoogle Scholar
- Pastan I. Targeted therapy of cancer with recombinant immunotoxins. Biochim Biophys Acta. 1997;1333:C1–6.PubMedGoogle Scholar
- Ghetie MA, Ghetie V, Vitetta ES. Immunotoxins for the treatment of B-cell lymphomas. Mol Med. 1997;3:420–7.PubMedGoogle Scholar
- Winkler U, Barth S, Schnell R, Diehl V, Engert A. The emerging role of immunotoxins in leukemia and lymphoma. Ann Oncol. 1997;8:139–46. DOIPubMedGoogle Scholar
- Murray LJ, Habeshaw JA, Wiels J, Greaves MF. Expression of Burkitt lymphoma-associated antigen (defined by the monoclonal antibody 38.13) on both normal and malignant germinal-centre B cells. Int J Cancer. 1985;36:561–5. DOIPubMedGoogle Scholar
- Taga S, Mangeney M, Tursz T, Wiels J. Differential regulation of glycosphingolipid biosynthesis in phenotypically distinct Burkitt's lymphoma cell lines. Int J Cancer. 1995;61:261–7. DOIPubMedGoogle Scholar
- LaCasse EC, Saleh MT, Patterson B, Minden MD, Gariepy J. Shiga-like toxin purges human lymphoma from bone marrow of severe combined immunodeficient mice. Blood. 1996;88:1551–67.PubMedGoogle Scholar
- Wheeler AH. Therapeutic uses of botulinum toxin. Am Fam Physician. 1997;55:541–8.PubMedGoogle Scholar
- Averbuch-Heller L, Leigh RJ. Medical treatments for abnormal eye movements: pharmacological, optical and immunological strategies. Aust N Z J Ophthalmol. 1997;25:7–13.PubMedGoogle Scholar
- Carter SR, Seiff SR. Cosmetic botulinum toxin injections. Int Ophthalmol Clin. 1997;37:69–79. DOIPubMedGoogle Scholar
- Maseri A, Andreotti F. Targeting new thrombolytic regimens at specific patient groups: implications for research and cost-containment. Eur Heart J. 1997;18:F28–35.PubMedGoogle Scholar
- Levine SR. Thrombolytic therapy for stroke: the new paradigm. Hosp Pract (Off Ed). 1997;32:57–73.
- Cherry JD. Comparative efficacy of acellular pertussis vaccines: an analysis of recent trials. Pediatr Infect Dis J. 1997;16:S90–6. DOIPubMedGoogle Scholar
- National Institutes of Health. The Jordan report: accelerated development of vaccines. 1998.
- Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–50. DOIGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 5, Number 2—April 1999
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Alison D. O'Brien, Uniformed Services University of the Health Sciences, Department of Microbiology and Immunology, 4301 Jones Bridge Road, Bethesda, MD 20814, USA; fax: 301-295-3773
Top