Volume 6, Number 6—December 2000
Synopsis
Hemophagocytic Syndromes and Infection
Cite This Article
Citation for Media
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an unusual syndrome characterized by fever, splenomegaly, jaundice, and the pathologic finding of hemophagocytosis (phagocytosis by macrophages of erythrocytes, leukocytes, platelets, and their precursors) in bone marrow and other tissues. HLH may be diagnosed in association with malignant, genetic, or autoimmune diseases but is also prominently linked with Epstein-Barr (EBV) virus infection. Hyperproduction of cytokines, including interferon-γ and tumor necrosis factor-α, by EBV-infected T lymphocytes may play a role in the pathogenesis of HLH. EBV-associated HLH may mimic T-cell lymphoma and is treated with cytotoxic chemotherapy, while hemophagocytic syndromes associated with nonviral pathogens often respond to treatment of the underlying infection.
Figure 1
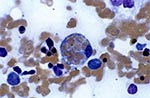
Figure 1. Hemophagocytosis in the bone marrow of an 18-year-old woman with Epstein-Barr virus (EBV)-associated hemophagocytic lymphohistiocytosis. The patient visited her physician in September 1997 with pharyngitis and an elevated heterophile agglutinin titer....
The term hemophagocytosis describes the pathologic finding of activated macrophages, engulfing erythrocytes, leukocytes, platelets, and their precursor cells (Figure 1) (1). This phenomenon is an important finding in patients with hemophagocytic syndrome, more properly referred to as hemophagocytic lymphohistiocytosis (HLH) (2). HLH is a distinct clinical entity characterized by fever, pancytopenia, splenomegaly, and hemophagocytosis in bone marrow, liver, or lymph nodes. The syndrome, which has also been referred to as histiocytic medullary reticulosis, was first described in 1939 (3). HLH was initially thought to be a sporadic disease caused by neoplastic proliferation of histiocytes. Subsequently, a familial form of the disease (4) (now referred to as familial hemophagocytic lymphohistiocytosis [5]) was described. However, the nearly simultaneous development of fatal HLH by a father and son in 1965 indicated that infection might play a role (6).
HLH has since been associated with a variety of viral, bacterial, fungal, and parasitic infections, as well as collagen-vascular diseases (7-11) and malignancies, particularly T-cell lymphomas (12-14). This diversity has prompted the suggestion that HLH secondary to an underlying medical illness should be designated reactive hemophagocytic syndrome. The association between HLH and infection is important because 1) both sporadic and familial cases of HLH are often precipitated by acute infections; 2) HLH may mimic infectious illnesses, such as overwhelming bacterial sepsis and leptospirosis (15); 3) HLH may obscure the diagnosis of a precipitating, treatable infectious illness (as reported for visceral leishmaniasis [16]); and 4) a better understanding of the pathophysiology of HLH may clarify the interactions between the immune system and infectious agents.
This article describes the clinical features and epidemiology of HLH and summarizes its association with infection; reviews evidence that this syndrome results from disordered cellular immunity; outlines options for treatment of patients with infection-associated HLH; and discusses issues related to hemophagocytosis in genetic, malignant, and autoimmune disease. The genetic basis of hemophagocytic syndromes has been reviewed in detail (17).
Clinical criteria for the diagnosis of HLH, proposed by the Histiocyte Society (2), include clinical, laboratory, and histopathologic features (Table). Fever and splenomegaly are the most common clinical signs, but hepatomegaly, lymphadenopathy, jaundice, and rash are also seen. The rash is commonly described as maculopapular, but nodular eruptions have also been described (22). Of central nervous system manifestations, encephalopathy, meningismus, and seizures are the most commonly reported (23,24). These clinical findings may suggest an acute viral infection, such as Epstein-Barr virus (EBV), cytomegalovirus (CMV) infection, viral hepatitis, or acute HIV seroconversion, a situation complicated by the association of these infections with HLH.
The most prominent laboratory abnormalities noted are cytopenias, which may be profound. Serum chemistry findings may suggest hemolysis, with hyperbilirubinemia and elevation of lactate dehydrogenase. Most patients have hypertriglyceridemia and marked elevation of ferritin (25,26). Serum fibrinogen is typically low, and there may be disseminated intravascular coagulation (18). Elevated circulating fibrin degradation products and serum ferritin in patients with HLH appear to be associated with increased risk for death (27).
Histopathologically, hemophagocytosis is seen in bone marrow, spleen, and lymph nodes (1,28) and occasionally the central nervous system (23,29) and skin (22). Activated macrophages may engulf erythrocytes, leukocytes, and platelets, their precursors, and cellular fragments. These cells appear "stuffed" with other blood cells. Hemophagocytosis may be present in the liver, but infiltration of the hepatic portal tracts with lymphocytes is also common (1,28).
HLH appears to affect all ages, although the hereditary and sporadic cases are reported primarily in children (30); a crude annual incidence of 1.2 cases of familial hemophagocytic lymphohistiocytosis per million children has been reported in Sweden (19). Large series of HLH cases have been reported in Hong Kong (18,31) and Taiwan (12,32), but whether the incidence of HLH is higher in Asia than in Europe or North America is not known. A seasonal pattern has been suggested in which cases may occur more often in the summer (32).
The familial form of HLH occurs in young children as a genetic disorder with autosomal recessive inheritance; possible loci for a responsible gene or genes have recently been mapped to the long arms of chromosomes 9 (33) and 10 (34). HLH may also occur as a complication of Chediak-Higashi syndrome (35) or after EBV infection in patients with X-linked lymphoproliferative syndrome (36). In these patients, fatal infectious mononucleosis may be pathologically indistinguishable from HLH (37).
In 1979, HLH was described in a cohort of patients who had serologic evidence of recent viral infections (38), and virus-associated hemophagocytic syndrome was proposed as a distinct clinical entity. Subsequently, HLH has been reported in association with a variety of infections, and the term reactive hemophagocytic syndrome has been suggested to distinguish HLH associated with an identifiable infectious or noninfectious etiology from its hereditary forms. However, the reactive and hereditary forms of the disease are difficult to distinguish; for example, patients with familial forms of HLH may have hemophagocytic syndrome after a documented viral infection (39).
Case reports and case series on the association of infections and HLH are summarized at URL: http://www.cdc.gov/ncidod/eid/vol6no6/fisman_refs.htm
Disseminated infection with an unusual organism in a patient with HLH may represent secondary infection in an immunocompromised host; however, the resolution of HLH following treatment of infection suggests that, in many cases, HLH is secondary to the underlying infection.
A diagnosis that takes into account all the underlying diseases associated with HLH would not be practical, and formal guidelines for evaluating patients with suspected infection-associated HLH have not been established. Nevertheless, all patients meeting the criteria for HLH should undergo initial diagnostic tests that include routine cultures of blood and urine and chest radiography to screen for such infections as miliary tuberculosis. Attempts should be made to screen for infection with EBV, CMV, and parvovirus B19, either through serologic testing or polymerase chain reaction, in-situ hybridization, or (in the case of CMV) immunofluorescent antigen testing. Serologic testing for HIV and human herpesvirus-6 infection should also be considered, and throat and rectal swabs should be taken for viral culture. Because of the association between HLH and fungal infections, lysis-centrifugation blood cultures and fungal antigen testing should be considered for all patients with HLH. Even if an infection known to be associated with HLH has been confirmed, cell marker and T-cell receptor gene rearrangement tests should be performed on bone marrow or other tissue specimens to determine whether an underlying T-cell lymphoma is present.
Extensive testing for underlying infecting organisms should be guided by epidemiologic data and the patient's medical history. For example, in a patient with underlying HIV infection, HLH has been associated with infections that commonly affect patients with AIDS (e.g., pneumococcal disease, pneumocystosis, histoplasmosis, and infection with Penicillium marneffei) and with T-cell lymphoma. Patients with a history of travel or animal exposure should be screened for such infections as leishmaniasis, brucellosis, rickettsioses, and malaria. In bone marrow transplant patients, attempts should be made to isolate adenovirus from urine, nasopharyngeal and rectal swabs, and tissue specimens.
Because so many immunologic, neoplastic, genetic, and infectious disorders may be associated with HLH, clinicians should work closely with pathologists and microbiologists to clearly define precipitating or underlying illnesses.
Phagocytosis of blood cells and their precursors is a hallmark of hemophagocytic syndromes. Hemophagocytosis is achieved mostly by monocytes, macrophages, and nitro-blue tetrazolium reduction by monocytes from patients with HLH is approximately six times that of control monocytes (40). Splenic macrophages from patients with HLH exhibit an activated phenotype with increased expression of MHC class I and II molecules and increased M-CSF receptor expression (41). Phagocytosis of platelets in HLH may be enhanced by increases in anti-platelet immunoglobulin (Ig) G, which has been reported in parvovirus B19-associated HLH (42).
Figure 2
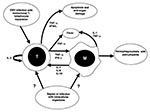
Figure 2. Schematic representation of possible immunopathologic mechanisms in infection-associated hemophagocytic lymphohistiocytosis (HLH). In Epstein-Barr virus (EBV)-associated HLH, infection of T lymphocytes results in clonal proliferation, with production of high levels of activating...
Excessive activation of monocytes in HLH may be due to stimulation by high levels of activating cytokines. High levels of interferon-γ (IFN-γ) (43-45), soluble interleukin-2 receptor (43,46), tumor necrosis factor-α (TNF-α) (44,47,48) interleukin-1 (49), and interleukin-6 (43) have been demonstrated, suggesting that elaboration of activating cytokines by T-helper cells promotes activation of macrophages in this disease (Figure 2). Higher levels of IFN-γ and TNF-α correlate with poor clinical outcome in children with virus-associated HLH (45,49).
Recently, oversecretion of interleukin-18 by monocytes in patients with HLH has been described (50); interleukin-18 production may further enhance TNF-γ and IFN-α production by T-lymphocytes and NK cells, as well as induce Fas ligand expression on lymphocytes, which enhances their cytotoxic effect. Serum levels of soluble Fas ligand, which can trigger apoptosis in such Fas-expressing tissues as the kidney, liver, and heart, also appear to be increased in HLH (51).
The exact mechanisms by which abnormal cytokine elaboration by T lymphocytes results in HLH remain unclear. However, data from patients with EBV-associated HLH, as well as HLH associated with EBV-positive T-cell lymphomas, may be instructive. Although T lymphocytes lack the putative EBV receptor CD21, the presence of episomal EBV genome in T-cell lymphomas (52,53) and T lymphocytes from patients with virus-associated HLH is well described (54,55). EBV-positive T-cell lymphomas appear to elaborate TNF-α more frequently than either EBV-positive B-cell lymphomas or EBV-negative T-cell lymphomas (53).
Lay and colleagues induced the expression of CD21 in T-lymphoma cell lines and subsequently infected these cells with EBV. High levels of TNF-α, IFN-γ, and IL-1α were secreted by these cells after EBV infection; when the lymphocytes were co-cultured with monocytes, enhanced phagocytosis by monocytes was observed. The enhanced phagocytosis was eliminated by the addition of antibodies against TNF-α and IFN-γ (53).
The clonal expansion of EBV-infected T lymphocytes has been demonstrated in both EBV-associated HLH (55-57) and EBV-positive T-cell lymphoma (53) by the presence of homogeneous viral terminal repetitive sequences. EBV-infected cells stain positive for such T-lymphocyte markers as CD45RO and T-cell receptor-β (54,55). Clonality of infected T lymphocytes is further suggested by the finding of monoclonal rearrangements of the T-cell receptor-β gene in EBV-associated HLH (58).
The distinction between the monoclonal proliferation of T lymphocytes seen in EBV-associated HLH and EBV-positive T-cell lymphomas may describe extremes of a spectrum of disordered T-lymphocyte proliferation and cytokine elaboration following EBV infection of T lymphocytes. Elaboration of such viral proteins as LMP1, essential to the immortalization of EBV-infected B-lymphocytes, may affect EBV infection of T lymphocytes, although studies suggest otherwise (59). It is also unclear whether clonal proliferation of T lymphocytes occurs in HLH associated with pathogens other than EBV. The fact that these syndromes seem more likely to resolve with control of the underlying infection suggests that this may not be the case. The apparent utility of cyclosporin A in HLH (60-62) and the morphologic similarity of the liver disease seen in HLH to acute graft rejection in transplant patients (1) lend further credence to the role of lymphocytes as central to the pathogenesis of HLH.
The pathophysiology of infection-associated HLH following infection with nonviral pathogens may also be related to production of high levels of activating cytokines by host lymphocytes and monocytes. The relative frequency of association between infecting organisms (e.g., Mycobacterium tuberculosis, Salmonella Typhi, and Leishmania sp.) that trigger a TH1 immune response and reactive hemophagocytic syndromes might suggest that the syndromes result from a poorly regulated or inappropriate TH1 response to intracellular pathogens. However, Tsuda and colleagues found no evidence of a marked shift towards a TH1 cytokine profile in patients with HLH associated with nonviral infections (63).
Because these disorders are rare, no controlled clinical trials of therapy have been performed. For patients with reactive HLH associated with pathogens other than EBV, supportive care and treatment of the underlying infection is associated with recovery in 60%-70% (20,64). Among adults with HLH, age > 30 years appears to be associated with an increased risk for death (27).
Epstein-Barr virus-associated HLH is almost universally fatal if untreated, with death usually resulting from hemorrhage, infection, or multiorgan failure (64,65). The poor prognosis of this syndrome suggests that patients should be treated initially with combination chemotherapy and immunotherapy, regardless of whether they are thought to have familial HLH. Chemotherapy with etoposide (which is toxic to macrophages) and dexamethasone is recommended, with the use of intrathecal methotrexate in patients with neurologic symptoms or persistent cerebrospinal fluid abnormalities (66-69). In a group of children with EBV-associated HLH, investigators induced complete remission (median 15 months) in 15 of 17 patients (68). The increasing recognition of the important role of T lymphocytes in HLH has led to the recommendation that chemotherapy be combined with cyclosporin A immunotherapy (60-62,67). Antithymocyte globulin may also have a role in therapy (60).
HLH associated with viral infection may be difficult to distinguish from familial HLH triggered by a viral infection (39), although familial HLH should be considered more likely in infants even in the absence of a positive family history (19). The distinction is important, as allogeneic bone marrow transplantation is the therapy of choice in patients with familial HLH who attain remission (67,70). In patients without a clear diagnosis of familial HLH, bone marrow transplantation should be considered if remission is not attained by 8 weeks of chemotherapy and immunotherapy. Patients in remission without a clear diagnosis of familial HLH should be monitored closely for signs of relapse (67).
The role of intravenous immunoglobulin in the treatment of HLH is unclear. Remission after such therapy has been reported in adults and older children with underlying immune dysfunction (71,72). However, Chen and colleagues noted remission in only two of nine children with virus-associated HLH treated with intravenous immunoglobulin alone (65).
Acyclovir does not appear to be useful in the treatment of EBV-associated HLH. However, resolution of HLH associated with other viral pathogens has been reported after antiviral chemotherapy. For example, adenovirus-associated HLH in a bone marrow transplant patient was reported to resolve with vidarabine (73), while human herpesvirus-8-associated HLH in a patient with HIV infection appeared to improve with the use of foscarnet (74).
HLH and related hemophagocytic syndromes are uncommon but severe illnesses associated with a variety of infectious agents, as well as genetic, neoplastic, and autoimmune diseases. HLH in the context of infection is best described as part of a spectrum of EBV-associated illness resulting in clonal proliferation of T-lymphocytes, with excessive activation of macrophages. This syndrome may be difficult to distinguish from T-cell lymphoma and should be treated aggressively with etoposide-based chemotherapeutic regimens.
Hemophagocytic syndromes associated with other infectious illnesses, including sepsis, typhoid fever, tuberculosis, and leishmaniasis, may resolve with treatment of the underlying infection, and their recognition is important as they may mimic malignant disease. Further study of these reactive hemophagocytic syndromes may yield important insights into the biology of macrophage activation.
Dr. Fisman is a physician in the Division of Infectious Diseases, Beth Israel Deaconess Medical, and a postdoctoral fellow in health policy at the Harvard Center for Risk Analysis. His research interests include mathematical modeling of infectious diseases and the assessment of cost-effectiveness of new diagnostic and therapeutic modalities for the treatment and control of infectious diseases.
Dr. Fisman is supported by a postdoctoral fellowship from the Agency for Healthcare Research and Quality.
Acknowledgment
The author thanks Margaret James Koziel for review of the manuscript and for many insightful comments.
References
- Favara BE. Hemophagocytic lymphohistiocytosis: a hemophagocytic syndrome. Semin Diagn Pathol. 1992;9:63–74.PubMedGoogle Scholar
- Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29–33.PubMedGoogle Scholar
- Scott R, Robb-Smith A. Histiocytic medullary reticulosis. Lancet. 1939;2:194–8.
- Farquhar J, Claireaux A. Familial hemophagocytic reticulosis. Arch Dis Child. 1952;27:519–25. DOIPubMedGoogle Scholar
- Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140:221–30. DOIPubMedGoogle Scholar
- Boake WC, Card WH, Kimmey JF. Histiocytic medullary reticulosis: concurrence in father and son. Arch Intern Med. 1965;116:245–52.PubMedGoogle Scholar
- Onishi R, Namiuchi S. Hemophagocytic syndrome in a patient with rheumatoid arthritis. Intern Med. 1994;33:607–11. DOIPubMedGoogle Scholar
- Wong KF, Hui PK, Chan JK, Chan YW, Ha SY. The acute lupus hemophagocytic syndrome. Ann Intern Med. 1991;114:387–90.PubMedGoogle Scholar
- Kumakura S, Ishikura H, Munemasa S, Adachi T, Murakawa Y, Kobayashi S. Adult onset Still's disease associated hemophagocytosis. J Rheumatol. 1997;24:1645–8.PubMedGoogle Scholar
- Morris JA, Adamson AR, Holt PJ, Davson J. Still's disease and the virus-associated haemophagocytic syndrome. Ann Rheum Dis. 1985;44:349–53. DOIPubMedGoogle Scholar
- Yasuda S, Tsutsumi A, Nakabayashi T, Horita T, Ichikawa K, Ieko M, Haemophagocytic syndrome in a patient with dermatomyositis. Br J Rheumatol. 1998;37:1357–8. DOIPubMedGoogle Scholar
- Chang CS, Wang CH, Su IJ, Chen YC, Shen MC. Hematophagic histiocytosis: a clinicopathologic analysis of 23 cases with special reference to the association with peripheral T-cell lymphoma. J Formos Med Assoc. 1994;93:421–8.PubMedGoogle Scholar
- Kadin ME, Kamoun M, Lamberg J. Erythrophagocytic T gamma lymphoma: a clinicopathologic entity resembling malignant histiocytosis. N Engl J Med. 1981;304:648–53. DOIPubMedGoogle Scholar
- Yao M, Cheng A, Su I. Clinicopathological spectrum of haemophagocytic syndrome in Epstein-Barr virus-associated peripheral T-cell lymphoma. Br J Haematol. 1994;87:535–43. DOIPubMedGoogle Scholar
- Yang CW, Pan MJ, Wu MS, Chen YM, Tsen YT, Lin CL, Leptospirosis: an ignored cause of acute renal failure in Taiwan. Am J Kidney Dis. 1997;30:840–5. DOIPubMedGoogle Scholar
- Matzner Y, Behar A, Beeri E, Gunders A, Hershko C. Systemic leishmaniasis mimicking malignant histiocytosis. Cancer. 1979;43:398–402. DOIPubMedGoogle Scholar
- Dufourcq-Lagelouse R, Pastural E, Barrat F, Feldmann J, Le Diest F, Fischer A, Genetic basis of hemophagocytic lymphohistiocytosis syndrome. Int J Mol Med. 1999;4:127–33.PubMedGoogle Scholar
- Wong KF, Chan J. Reactive hemophagocytic syndrome: a clinicopathologic study of 40 patients in an Oriental population. Am J Med. 1992;93:177–80. DOIPubMedGoogle Scholar
- Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80:428–35. DOIPubMedGoogle Scholar
- Reiner AP, Spivak J. Hemophagocytic histiocytosis: a report of 23 new patients and a review of the literature. Medicine. 1988;67:369–88.PubMedGoogle Scholar
- Sailler L, Duchayne E, Marchou B, Brousset P, Pris J, Massip P, Aspects etiologiques des hemophagocytoses reactionnelles: etude retrospective chez 99 patients. Rev Med Interne. 1997;18:855–64. DOIPubMedGoogle Scholar
- Smith KJ, Skelton H, Yeager J, Angritt P, Wagner K, James W, Military Medical Consortium for Applied Retroviral Research. Cutaneous histopathologic, immunohistochemical, and clinical manifestations in patients with hemophagocytic syndrome. Arch Dermatol. 1992;128:193–200. DOIPubMedGoogle Scholar
- Henter JT, Nennesmo I. Neuropathologic findings and neurologic symptoms in twenty-three children with hemophagocytic lymphohistiocytosis. J Pediatr. 1997;130:358–65. DOIPubMedGoogle Scholar
- Haddad E, Sulis ML, Jabado N, Blanche S, Fischer A, Tardieu M. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997;89:794–800.PubMedGoogle Scholar
- Koduri PR, Carandang G, DeMarais P, Patel AR. Hyperferritinemia in reactive hemophagocytic syndrome: report of four adult cases. Am J Hematol. 1995;49:247–9. DOIPubMedGoogle Scholar
- Esumi N, Ikushima S, Hibi S, Todo S, Imashuku S. High serum ferritin level as a marker of malignant histiocytosis and virus-associated hemophagocytic syndrome. Cancer. 1987;61:2071–6. DOIPubMedGoogle Scholar
- Kaito K, Kobayashi M, Katayama T, Otsubo H, Ogasawara Y, Sekita T, Prognostic factors in hemophagocytic syndrome in adults: analysis of 34 cases. Eur J Haematol. 1997;59:247–53. DOIPubMedGoogle Scholar
- Ost A, Nilsson-Ardnor S, Henter J. Autopsy findings in 27 children with haemophagocytic lymphohistiocytosis. Histopathology. 1998;32:310–6. DOIPubMedGoogle Scholar
- Martin JJ, Cras P. Familial erythrophagocytic lymphohistiocytosis: a neuropathological study. Acta Neuropathol. 1985;66:140–4. DOIPubMedGoogle Scholar
- Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203.PubMedGoogle Scholar
- Wong KF, Chan JK, Lo ES, Wong CS. A study of the possible etiologic association of Epstein-Barr virus with reactive hemophagocytic syndrome in Hong Kong Chinese. Hum Pathol. 1996;27:1239–42. DOIPubMedGoogle Scholar
- Chen RL, Su IJ, Lin KH, Lee SH, Lin DT, Chu WM, Fulminant childhood hemophagocytic syndrome mimicking histiocytic medullary reticulosis. An atypical form of Epstein-Barr virus infection. Am J Clin Pathol. 1991;96:171–6.PubMedGoogle Scholar
- Ohadi M, Lalloz MR, Sham P, Zhao J, Dearlove AM, Shiach C, Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64:165–71. DOIPubMedGoogle Scholar
- Dufourcq-Lagelouse R, Jabado N, Le Diest F, Stephan J, Souillet G, Bruin M, Linkage of familial hemophagocytic lymphohistiocytosis to 10q21-22 and evidence for heterogeneity. Am J Hum Genet. 1999;64:172–9. DOIPubMedGoogle Scholar
- Rubin CM, Burke B, McKenna R, McClain K, White J, Nesbit M, The accelerated phase of Chediak-Higashi syndrome: an expression of the virus-associated hemophagocytic syndrome? Cancer. 1984;56:524–30. DOIPubMedGoogle Scholar
- Purtilo DT, DeFlorio D Jr, Hutt LM, Bhawan J, Yang JP, Otto R, Variable phenotypic expression of an X-linked recessive lymphoproliferative syndrome. N Engl J Med. 1977;297:1077–80. DOIPubMedGoogle Scholar
- Mroczek EC, Weisenburger D, Grierson H, Markin R, Purtilo D. Fatal infectious mononucleosis and virus-associated hemophagocytic syndrome. Arch Pathol Lab Med. 1987;111:530–5.PubMedGoogle Scholar
- Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr, Simmons RL, Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979;44:993–1002. DOIPubMedGoogle Scholar
- Henter JT, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82:369–72. DOIPubMedGoogle Scholar
- Burgio GR, Arico M, Marconi M, Lanfranchi A, Caselli D, Ugazio AG. Spontaneous NBT reduction by monocytes as a marker of disease activity in children with histiocytosis. Br J Haematol. 1990;74:146–50. DOIPubMedGoogle Scholar
- Ker éveur A, McIlroy D, Samri A, Oksenhendler E, Clauvel JP, Autran B. Up-regulation of adhesion and MHC molecules on splenic monocyte/macrophages in adult haemophagocytic syndrome. Br J Haematol. 1999;104:871–7. DOIPubMedGoogle Scholar
- Toyoshige M, Takahashi H. Increase of platelet-associated IgG (PA-IgG) and hemophagocytosis of neutrophils and platelets in parvovirus B19 infection. Int J Hematol. 1998;67:205–6. DOIPubMedGoogle Scholar
- Fujiwara F, Hibi S, Imashuku S. Hypercytokinemia in hemophagocytic syndrome. Am J Pediatr Hematol Oncol. 1993;15:92–8. DOIPubMedGoogle Scholar
- Ohga S, Matsuzaki A, Nishizaki M, Nagashima T, Kai T, Suda M, Inflammatory cytokines in virus-associated hemophagocytic syndrome: interferon gamma as a sensitive indicator of disease activity. Am J Pediatr Hematol Oncol. 1993;15:291–8.PubMedGoogle Scholar
- Imashuku S, Hibi S, Fujiwara F, Ikushima S, Todo S. Haemophagocytic lymphohistiocytosis, interferon gamma-nemia and Epstein-Barr virus involvement. Br J Haematol. 1994;88:656–8. DOIPubMedGoogle Scholar
- Komp DM, McNamara J, Buckley P. Elevated soluble interleukin-2 receptor in childhood hemophagocytic histiocytic syndromes. Blood. 1989;73:2128–32.PubMedGoogle Scholar
- Watanabe M, Shimamoto Y, Yamaguchi M, Inada S, Miyazaki S, Sato H. Viral-associated haemophagocytosis and elevated serum TNF-alpha with parvovirus-B19-related pancytopenia in patients with hereditary spherocytosis. Clin Lab Haematol. 1994;16:179–82. DOIPubMedGoogle Scholar
- Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–3.PubMedGoogle Scholar
- Ishii E, Ohga S, Aoki T, Yamada S, Sako M, Tasaka H, Prognosis of children with virus-associated hemophagocytic syndrome and malignant histiocytosis: correlation with levels of serum interleukin-1 and tumor necrosis factor. Acta Haematol. 1991;85:93–9. DOIPubMedGoogle Scholar
- Takada H, Ohga S, Mizuno Y, Suminoe A, Matsuzaki A, Ihara K, Oversecretion of IL-18 in haemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br J Haematol. 1999;106:182–9. DOIPubMedGoogle Scholar
- Hasegawa D, Kojima S, Tatsumi E, Hayakawa A, Kosaka Y, Nakamura H, Elevation of the serum Fas ligand in patients with hemophagocytic syndrome and Diamond-Blackfan anemia. Blood. 1998;91:2793–9.PubMedGoogle Scholar
- Su IJ, Hsu YH, Lin MT, Cheng AL, Wang CH, Weiss LM. Epstein-Barr virus-containing T-cell lymphoma presents with hemophagocytic syndrome mimicking malignant histiocytosis. Cancer. 1993;72:2019–27. DOIPubMedGoogle Scholar
- Lay JD, Tsao CJ, Chen JY, Kadin ME, Su IJ. Upregulation of tumor necrosis factor-alpha gene by Epstein-Barr virus and activation of macrophages in Epstein-Barr virus-infected T cells in the pathogenesis of hemophagocytic syndrome. J Clin Invest. 1997;100:1969–79. DOIPubMedGoogle Scholar
- Su IJ, Chen R, Lin D, Lin K, Chen C. Epstein-Barr virus (EBV) infects T lymphocytes in childhood EBV-associated hemophagocytic syndrome in Taiwan. Am J Pathol. 1994;144:1219–25.PubMedGoogle Scholar
- Kawaguchi H, Miyashita T, Herbst H, Niedobitek G, Asada M, Tsuchida M, Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus- associated hemophagocytic syndrome. J Clin Invest. 1993;92:1444–50. DOIPubMedGoogle Scholar
- Mori M, Kurozumi H, Akagi K, Tanaka Y, Imai S, Osato T. Monoclonal proliferation of T cells containing Epstein-Barr virus in fatal mononucleosis. N Engl J Med. 1992;327:58. DOIPubMedGoogle Scholar
- Chen JS, Tzeng CC, Tsao CJ, Su WC, Chen TY, Jung YC, Clonal karyotype abnormalities in EBV-associated hemophagocytic syndrome. Haematologica. 1997;82:572–6.PubMedGoogle Scholar
- Craig FE, Clare N, Sklar J, Banks P. T-cell lymphoma and the virus-associated hemophagocytic syndrome. Am J Clin Pathol. 1991;97:189–94.PubMedGoogle Scholar
- Ohshima K, Suzumiya J, Sugihara M, Nagafuchi S, Ohga S, Kikuchi M. Clinicopathological study of severe chronic active Epstein-Barr virus infection that developed in association with lymphoproliferative disorder and/or hemophagocytic syndrome. Pathol Int. 1998;48:934–43. DOIPubMedGoogle Scholar
- St éphan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993;82:2319–23.PubMedGoogle Scholar
- Oyama Y, Amano T, Hirakawa S, Hironaka K, Suzuki S, Ota Z. Haemophagocytic syndrome treated with cyclosporin A: a T cell disorder? Br J Haematol. 1989;73:276–8. DOIPubMedGoogle Scholar
- Tsuda H, Shirono K. Successful treatment of virus-associated haemophagocytic syndrome in adults by cyclosporine A supported by granulocyte colony-stimulating factor. Br J Haematol. 1996;93:572–4. DOIPubMedGoogle Scholar
- Tsuda H, Fujisao S. Th1/Th2 milieu in adult hemophagocytic syndrome. Acta Haematol. 1999;101:157–60. DOIPubMedGoogle Scholar
- Janka G, Imashuku S, Elinder G, Schneider M, Henter JI. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:435–44. DOIPubMedGoogle Scholar
- Chen RL, Lin KH, Lin DT, Su IJ, Huang LM, Lee PI, Immunomodulation treatment for childhood virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol. 1995;89:282–90. DOIPubMedGoogle Scholar
- Imashuku S, Hibi S, Ohara T, Iwai A, Sako M, Kato M, Effective control of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis with immunochemotherapy. Blood. 1999;93:1869–74.PubMedGoogle Scholar
- Henter JI, Arico M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:417–33. DOIPubMedGoogle Scholar
- Imashuku S, Hibi S, Ohara T, Iwai A, Sako M, Kato M, Effective control of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis with immunochemotherapy. Blood. 1999;93:1869–74.PubMedGoogle Scholar
- Chen JS, Lin K, Lin D, Chen R, Jou S, Su I. Longitudinal observation and outcome of nonfamilial childhood haemophagocytic syndrome receiving etoposide-containing regimens. Br J Haematol. 1998;103:756–62. DOIPubMedGoogle Scholar
- Blanche S, Caniglia M, Girault D, Landman J, Griscelli C, Fischer A. Treatment of hemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation: a single-center study of 22 cases. Blood. 1991;78:51–4.PubMedGoogle Scholar
- Koch WC, Massey G, Russell CE, Adler SP. Manifestations and treatment of human parvovirus B19 infection in immunocompromised patients. J Pediatr. 1990;116:355–9. DOIPubMedGoogle Scholar
- Gill DS, Spencer A, Cobcroft R. High-dose gamma-globulin therapy in the reactive haemophagocytic syndrome. Br J Haematol. 1994;88:204–6. DOIPubMedGoogle Scholar
- Kitabayashi A, Hirokawa M, Kuroki J, Nishinari T, Niitsu H, Miura AB. Successful vidarabine therapy for adenovirus type 11-associated acute hemorrhagic cystitis after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1994;14:853–4.PubMedGoogle Scholar
- L öw P, Neipel F, Rascu A, Steininger H, Manger B, Fleckenstein B, Suppression of HHV-8 viremia by foscarnet in an HIV-infected patient with Kaposi's sarcoma and HHV-8 associated hemophagocytic syndrome. Eur J Med Res. 1998;3:461–4.PubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 6, Number 6—December 2000
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
David Fisman, Division of Infectious Diseases, Kennedy-6, Beth Israel Deaconess Medical Center, 1 Autumn Street, Boston, MA 02215; Fax: (617)-632-0766
Top