Volume 9, Number 7—July 2003
Research
Mutations in Putative Mutator Genes of Mycobacterium tuberculosis Strains of the W-Beijing Family
Cite This Article
Citation for Media
Abstract
Alterations in genes involved in the repair of DNA mutations (mut genes) result in an increased mutation frequency and better adaptability of the bacterium to stressful conditions. W-Beijing genotype strains displayed unique missense alterations in three putative mut genes, including two of the mutT type (Rv3908 and mutT2) and ogt. These polymorphisms were found to be characteristic and unique to W-Beijing phylogenetic lineage. Analysis of the mut genes in 55 representative W-Beijing isolates suggests a sequential acquisition of the mutations, elucidating a plausible pathway of the molecular evolution of this clonal family. The acquisition of mut genes may explain in part the ability of the isolates of W-Beijing type to rapidly adapt to their environment.
Tuberculosis (TB) and AIDS cause more deaths in adults worldwide than any other infectious disease. Globally, the number of TB cases is growing at a rate of 2% per year. Resistance, especially multidrug-resistance (MDR), is an increasing problem (1) and a growing hazard to human health. Many outbreaks of MDR-TB, defined as resistance to at least rifampicin and isoniazid, have been reported, with poor response to therapy and very high disease and death rates. Some TB outbreaks have involved patients with HIV co-infection (2,3). Although in several instances, MDR outbreaks associated with a particular genotype, such as the W strain, have been identified (4,5), drug-susceptible variants of the W strain account for most of this group of isolates characterized to date.
Figure 1
Figure 1. Characteristic patterns of Mycobacterium tuberculosis Beijing genotype strains.
In 1995, the largest proportion of the Mycobacterium tuberculosis strains from Beijing, China, shared a high degree of similarity in IS6110 restriction fragment length polymorphism (RFLP) patterns and identical spoligo patterns (6). Subsequent molecular analyses have indicated that the W and Beijing isolates constitute a single group of strains designated as the W-Beijing genotype (Figure 1). The global distribution and success of M. tuberculosis isolates of the W-Beijing genotype have led to the hypothesis that these strains may have selective advantages over other M. tuberculosis strains. In addition to the W-MDR strain identified in New York City, and areas in Cuba, Estonia, Vietnam, and Russia, the W-Beijing genotype has been significantly associated with drug resistance (7 and unpub. data). Several studies have suggested that the W-Beijing genotype strains are disseminating throughout the world (7). In Vietnam, the proportion of W-Beijing strains was 71% in patients <25 years of age and 41% for those >25 years of age (8). Furthermore, W-Beijing strains have been implicated in several TB epidemics globally, including ones in New York, Texas, California, South Carolina, and New Jersey in the United States (9) and South Africa, Russia, and Spain (10). A recent study showed that 82% of MDR strains isolated in a prison in Azerbaijan, Eastern Europe, are of the W-Beijing genotype (11).
Ongoing research is focused on identifying the factors responsible for the worldwide spread of the W-Beijing strains and their ability to adapt and enhance their pathogenicity or virulence. Identifying a possible mechanism for increased adaptation of these bacteria to the human immunologic host defense system or human interventions such as anti-TB treatment is of the utmost importance. Such mechanisms may indicate how the bacterium adapts to the host, a prerequisite for an enhanced accumulation of genomic mutations associated with resistance. In M. tuberculosis, resistance to antibiotics occurs because of genomic mutations in certain genes, such as the katG gene for isoniazid (INH) resistance and the rpoB gene for rifampicin resistance (12). In contrast to several other pathogens with MDR phenotypes, plasmid or transposon-mediated mechanisms of resistance have not been reported in M. tuberculosis (13–15). Since resistance to bacteriostatic in M. tuberculosis is exclusively due to genomic mutations, the bacterium would benefit from an increased mutation rate.
Recent studies provided evidence for a role of mutator phenotypes in the emergence of MDR clinical Pseudomonas isolates (16). Such phenotypes not only enable the bacteria to acquire resistance to antibiotics more easily but also facilitate their adaptation to a new niche. Bacteria can escape immune surveillance by modulating bacterial resistance to host defense mechanisms (16–18). This finding prompted us to investigate whether a similar situation exists in M. tuberculosis. We have undertaken a comprehensive comparative sequence analysis of selected target genes to evaluate and study the presence of mutations in putative genes expected to play a role in the mutation frequency in such strains.
Mutated phenotypes commonly result from defects in DNA repair (19). An in silico analysis suggested that most mismatch repair systems (e.g., mutS, mutL, or mutH) were missing in the M. tuberculosis genome (20). However, the frequency of spontaneous mutations in M. tuberculosis (in vitro cultures) is similar to that found in other bacteria-carrying mismatch repair systems (21), which suggests that other DNA repair mechanisms must be present. Hypothetical open reading frames (ORF), similar to genes known to be responsible for the avoidance or repair of DNA lesions resulting from the alkylation or oxidation of nucleotides, are present in the genome of M. tuberculosis. We searched for variations in these genes in 139 clinical isolates to detect possible mutations that could allow an enhanced adaptability to the host and increased resistance to anti-TB drugs.
We searched for mut genes variation in 139 M. tuberculosis complex strains originating from 35 different countries. Ninety-four of these strains were selected because they were representative strains characterized with 13 different genetic markers in previous studies (6,22).
This set comprised 125 M. tuberculosis strains, 1 M. africanum, 8 M. bovis, 3 M. bovis BCG, and 2 M. microti. Fifty-five strains had a W-Beijing genotype; 12 had an MDR phenotype. Strains representing different branches of the W-Beijing genotype were studied. Eight MDR M. tuberculosis strains with a genotype other than Beijing were included. Five M. tuberculosis strains of the W-Beijing genotype and three strains of unrelated genotype were obtained from the national program for surveillance of MDR tuberculosis in Spain. Four M. tuberculosis W-Beijing genotype strains isolated in the Netherlands and one from Vietnam were included because they showed spoligo patterns with fewer than nine spacers. Five other W-Beijing genotype strains showed hybridization to an additional spacer, as demonstrated by using the extended set of spacers, two of which lacked hybridization to spacer 37. Strain W4 is part of a drug-susceptible outbreak in New Jersey (4); W147 is a drug-resistant isolate widely spread in Russia (23). Eleven strains were representative of ancestral W-Beijing strains, which diverged early in the evolution of the W-Beijing phylogenetic lineage. Finally, 29 strains of another frequently observed genotype, the Haarlem genotype (6), were investigated.
The collection consisted of 55 W-Beijing genotype isolates, 29 Haarlem genotype isolates, 8 strains of the African genotype, 1 M. bovis strain, and 46 representatives of other genotypes. Principal genetic grouping (PGG), according to the polymorphism in katG and gyrA, was known (24) for most of the isolates in this study. Seventy-four strains belong to PGG 1, 54 to PGG 2, and 3 to PGG 3. All isolates were subjected to at least IS6110 RFLP typing and spoligotyping (6). Drug susceptibility was determined for 41 of 139 strains (Table 1 and 2). Several putative mut genes were annotated as such in the released genome sequence of M. tuberculosis (25). In addition, using the BLAST program (26), we identified Rv3908 as an ORF carrying a mutT domain (27) and have since named it mutT4.
Primers were designed to amplify putative mut genes: mutY (5′-CCGGCGACGAATCGCTCGTT-3′, 5′-AGCTGGGACAGTCGTCGCGG-3′), mutM (5′-CTGGTTCGATGGTGATGACC-5′, 5′-GTGCGCTCGACCCACAG-3′), mutT2 (5′-TCCGGATGATGATTTACCTCC-3′, 5′-TCCGCCGGGTCGGGGAC-3′), mutT1 (5′-ATCGTCGGCGTGCCGTG-3′, 5′-GTCAGCGTCCTGCCCGG-3′), mutT4 (5′-TCGAAGGTGGGCAAATCGTG-3′ 5′-TGGGGTTCGCTGGAAGTGG–3′), ogt (5′-CAGCGCTCGCTGGCGCC-3′, 5′-GACTCAGCCGCTCGCGA-3′), and mut T3 (5′-GTCACGTCTGTTAGGACCTC-3′, 5′-CGCGCAACGGCTGCCGG-3′). Similar primers were designed to amplify the rpoB gene (5′-TACGGTCGGCGAGCTGATCC-3′, 5′- TACGGCGTTTCGATGAACC-3′).
DNA sequencing was performed directly on the amplified fragments by using the dideoxy chain-termination method with the Big Dye Terminator Cycle sequencing Kit (Perkin Elmer Applied Biosystems, Courtaboeuf, France) on a GeneAmp polymerase chain reaction (PCR) system 9600 (Perkin Elmer) and run on a DNA analysis system model 373 or 3100 (Applied Biosystems). Sequences of mutY, mutT2, mutT4, rpoB, mutT1, mutT3, and ogt of the M. tuberculosis strains H37Rv, CDC1551, and MT210 were obtained from published sequences or at the TIGR Web site (available from: URL: http://www.tigr.org/).
We searched for allele variation in putative genes coding for DNA repair enzymes: mutT (which hydrolyzes 8-oxo-deoxyguanosine triphosphate) (28), ogt (which removes methyl groups from O6-methylguanine in DNA) (29), mutM (formamidopyrimidine-DNA glycosylate) (30), and mutY (specific adenine glycosylate) (31) in 12 MDR M. tuberculosis strains. Subsequently, we genotyped for the observed single nucleotide polymorphism (SNP) variation in 124 strains, members of the M. tuberculosis complex, and in the three published sequences of M. tuberculosis strains. In total, the sampling comprises 55 W-Beijing genotype M. tuberculosis strains, including 11 ancestral W-Beijing isolates (unpub. data ), 84 M. tuberculosis strains of other genotypes, and 1 M. bovis strain.
Figure 2
Figure 2. MutT proteins’ sequences alignment. Mycobacterium tuberculosis Rv2985(MutT1), Rv1160(MutT2), Rv0413(MutT3), and Rv3908(MutT4) were selected from the M. tuberculosis genome because of their annotation or after a BLAST analysis. These sequences were compared...
Several putative mut genes were annotated as such in the released genome sequence of M. tuberculosis. A BLAST search using the E. coli mutT sequences as template identified, in addition to mutT1, mutT2, mutT3, the hypothetical ORF Rv3908, which we have designated as mutT4. The best matches with E. coli mutT gene were observed for mutT2 and mutT4. Figure 2 depicts sequence alignment of the conserved region of the different genes of the M. tuberculosis genome showing similarity with mutT of E. coli. The search for sequences similar to ogt, mutM, and mutY identified a single ORF in each case. Primers were designed for PCR amplification of all the genes mentioned above.
We first determined the sequences of the different genes mentioned above in 12 MDR M. tuberculosis strains (ZA20, ZA65, ZA67, ZA68, ZA69, ZA11, ZA16, ZA12, ZA13, ZAA14, ZA17, and ZA19), including 5 W-Beijing strains (ZA20, ZA65, ZA67, ZA78, and ZA69). For the mutY, mutM, mutT1, and mutT3 putative genes, PCR amplification was obtained in all strains tested, but sequence analysis did not indicate any nucleotide changes at these loci except for the same silent SNP in mutT3 in strains with a Haarlem genotype. We confirmed these findings by sequencing mutT1, mutT3, mutM, and mutY in a collection of 26 MDR strains from North Africa. No variation was observed in mutT1 or mutM. Only one strain had a major variation in mutY. All Haarlem strains carried one characteristic silent mutation in mutT3 and one characteristic mutation in ogt (Ser 15 replaced by Thr). These defining SNPs were also observed for all Haarlem strains of this study. No other variations were observed in mutT1, mutT3, mutM, or mutY. However, comparative sequence analysis of H37Rv, CDC1551, and the five MDR–W-Beijing isolates indicated polymorphisms in mutT2, mutT4, and ogt. These mutations in mutT4, mutT2, and ogt were also found in the W-Beijing strain 210 (TIGR) but not in MDR strains other than those belonging to the W-Beijing genotype. We therefore decided to extend this investigation and look for mutations in these three genes in a collection of M. tuberculosis complex isolates, including well-defined branches of the W-Beijing phylogenetic lineage (Table 1 and 2).
In 43 of 55 strains with a W-Beijing genotype, either susceptible to bacteriostat or MDR, we found a mutation in mutT4. Codon 48 (CGG) of the annotated ORF had been changed to GGG, resulting in the amino acid substitution of Arg by Gly (Table 1 and 2). All 11 W-Beijing isolates known to be closely related to the ancestral W-Beijing strain (AM, HI, N16, DU2, DV, LB2, KY, IK, 122(C11), 113, and 107(LB)) were found to have the wild-type genotype as all other 84 isolates with a genotype other than W-Beijing.
Thirty-nine of 43 W-Beijing strains with the mutation in mutT4 carried an additional mutation in mutT2 and in ogt. The mutT2 mutation constitutes a change in codon 58 (GGA to CGA), resulting in an amino acid substitution of Gly by Arg. The active site of the E. coli MutT enzyme comprises amino acids 53, 56, 57, and 98. Therefore, a mutation Gly to Arg at position 58 may have a important effect on enzyme activity and lead to a mutator phenotype.
All 39 W-Beijing isolates carrying the mutT2 polymorphism at codon 58 also displayed a concurrent silent mutation in codon 12 (Gly GGG to GGA Gly) of the ogt gene. Of four possible codons encoding for glycine, GGG and GGA had the lowest relative synonymous codon usage (RSCU) in genes with high expression levels (0.20 and 0.17, respectively, compared to 1.32 and 2.31 for GGU and GGC). For genes with low expression levels, the RSCU values are 0.92, 0.37, 0.65, and 2.06 for GGG, GGA, GGU, and GGC, respectively (32).
Figure 3
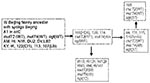
Figure 3. Schematic representation of a plausible pathway to explain the accumulation of mutations in mut genes.
The five W-Beijing isolates with a mutation in mutT4 and a wild-type mutT2 gene did not contain the ogt silent mutation on codon 12 either. Instead, they all shared a dinucleotide substitution in codon 37 (ACC to CTC) of ogt, resulting in amino acid substitution of Arg to Leu. These five W-Beijing isolates of 43 with the mutT4 mutations, without the mutT2 (codon 58) or the ogt (codon 12) mutations, differed molecularly from all other W-Beijing isolates in their spoligotype pattern and accompanying deletion flanking the DR locus. Four of five were isolated from Dutch patients in the Netherlands; the fifth originated from a patient in Vietnam. The Vietnamese isolate (no. 94) shared >95% IS6110 pattern similarity with the Dutch isolate 115 when standard RFLP analysis was used. Overall, the five isolates were closely related to each other according to IS6110 profiling (>90% similarity). Spacer 37 in the DR locus of Dutch isolates 114 and 139 was absent, while sample 115 was missing spacers 37 and 38, and 111 had a deletion of spacers 38 and 39 but not spacer 37, suggesting that these isolates may belong to a different sublineage. A tentative phylogeny of the W-Beijing strains analyzed in this study is proposed in Figure 3. Seven of nine MDR W-Beijing strains carried missense mutations in two muT genes (mutT2 and mutT4), and two had a missense mutation in both mutT4 and ogt (Table 1 and 2).
No mutations in mutT4 or in mutT2 were observed in any of the 84 M. tuberculosis complex strains, including 19 strains of PGG1, 54 strains of PGG2, and 2 strains of PGG3; the strains originated from 29 countries and were a genotype other than W-Beijing. A Thr15Ser mutation was observed in 24 of 29 strains of the Harlem family. No other change was observed in ogt.
Resistance to rifampicin in MDR strains was correlated with mutations in the rpoB gene. The three tested MDR W-Beijing strains isolated in Spain, with the mutations at the mutT2 and mutT4 loci, harbored a different mutation in the rpoB gene. These strains were isolated from patients who had emigrated from Eastern Europe to Spain (ZA67, ZA68, and ZA69). Analysis of the IS6110 RFLP of the respective isolates showed a difference of a single band. These findings suggest that the three strains may be related. The acquisition of the three different mutations in the rpoB gene leading to rifampicin resistance must have occurred after the acquisition of mutations in the putative nucleotide repair enzyme genes mutT4 and mutT2.
Our results show that M. tuberculosis strains of the W-Beijing genotype acquired missense mutations in DNA repair genes. These M. tuberculosis W-Beijing genotype strains are genetically highly conserved and widespread. DNA repair genes have been previously shown to be associated with mutator phenotypes in other microorganisms. The success of this group of strains may result in part from mutations in DNA repair enzymes, which might provided a true selective advantage for these bacteria to adapt and persist, including through the acquisition of resistance to anti-TB drugs. Mutations in the DNA repair genes might be the evolutionary answer of the TB bacillus to increase adaptation to hosts. This adaptation will lead to increasing trends in the TB epidemic in the coming decades. The World Health Organization considers MDR and resistance as a problem of local rather than of global importance (1). If the relative contribution of W-Beijing genotype strains to the current worldwide TB epidemic is increasing as suggested (7), this approach should be revised. In areas with an increasing problem with MDR-TB, such as Estonia and Russia, W-Beijing genotype strains are predominantly associated with MDR cases (33). In Germany, the relative proportion of W-Beijing strains among isolates from resistant cases has increased from 12% in 1995 to 35% in 2000 (unpub. data). The latter observation may indicate an increasing influence of W-Beijing strains on the worldwide TB epidemic.
We identify polymorphisms in M. tuberculosis in genes that might result in a mutator phenotype and therefore a plausibly better adaptation of the bacilli to a hostile environment (34). Forty-three of 55 W-Beijing isolates analyzed were found to have a unique mutation on the ORF Rv3908. This ORF contains a MutT domain and is denoted here as mutT4. Thirty-nine of 43 W-Beijing strains carried an additional and identical mutation in a second putative gene of the mutT family, mutT2, and an identical silent mutation in ogt.
The W-Beijing phylogenetic lineage probably acquired the mutation on codon 48 of the mutT4 only once and before other mutations associated with the mutator genes we describe. This mutation clearly distinguishes ancestral W-Beijing isolates from contemporary W-Beijing strains. The 11 W-Beijing isolates that did not have the characteristic mutT4 mutation on codon 48, consist of a collection of isolates known to be ancestral within this phylogenetic lineage, as determined by various other molecular techniques (unpub. data).
Nine of W-Beijing strains with the wild-type mutT2 gene had a characteristic mutation on codon 37 of the ogt gene, which suggests that these isolates constitute a branch of the W-Beijing family that diverged after the acquisition of the mutT4 mutation but before the development of the nucleotide substitution on mutT2. One strain carries the mutation 37 in ogt but no mutation in mutT4, a reversion that might have occurred after a transient mutator phenotype.
A mutation in mutT2 was always associated with a mutation in muT4. A first mutation may have occurred in mutT4 and thereafter a second mutation either in mutT2 or ogt was acquired. As observed for other bacterial populations, mutator phenotypes may be transient in many cases to limit deleterious effects (35). Identifying these mutations may aid in the identification of mut genes in M. tuberculosis. These mutations associated with mutator genes provide a reliable tool for the identification of W-Beijing isolates and thus a useful marker for strains endowed with capacity to yield epidemics. The biologic consequences of these mutations and function of these DNA repair genes are currently been investigated in the laboratory.
Nine MDR strains with a W-Beijing genotype were among strains carrying two missense mutations in putative mutator genes. Phylogenetically unrelated M. tuberculosis MDR isolates had no mutations within the DNA repair genes investigated in this study. Our data support the idea that M. tuberculosis strains of the W-Beijing genotype may have adapted to hostile environments, including exposure to anti-TB drugs, because of a succession of alterations of DNA repair enzymes. Other genes involved in other DNA repair mechanisms or in the fidelity of DNA replication may also be involved and remain to be investigated.
The acquisition of mutator alleles was described as an adaptive response of bacteria to a succession of different environments (18,35,36). After infecting a host, M. tuberculosis has to adapt to different environments such as alveolar macrophages and dendritic cells and subsequently to granuloma containing inactivated macrophages or to activated macrophages after induction of the acquired immune responses. In addition, the bacilli have to adapt to the caseous media with low oxygen concentration in the center of tubercles and to different types of tissues during dissemination of the disease. Such variable growth conditions might select for mutations in M. tuberculosis strains, as described in other bacterial populations exposed to different environmental challenges. Mutations and selection might occur with an increased frequency caused by the toxic radicals produced in phagocytic cells.
However, a mutator phenotype is often transient. Otherwise a continual accumulation of mutations would lead to deleterious effects and loss of fitness. No difference in the frequency of spontaneous mutations, resulting in a rifampin resistance phenotype, was observed for W-Beijing strains (37). We suggest that a transient mutator phenotype allowed a better adaptation of W-Beijing strains. Subsequent compensatory mutations occurred to reverse the mutator phenotype. An alternative hypothesis would be the existence of a higher mutation rate in specific conditions (i.e., in mutagenic radicals inside phagocytes). The accumulation of mutations leading to antibiotic resistance in W-Beijing strains may be a consequence of the appearance of strains with a better adaptation to the hosts. MDR strains would be easily selected when patients with strains that have adapted better received inadequate anti-TB regimens.
Dr. Rad is a molecular biologist. Her expertise includes DNA amplification, sequencing, and cloning.
Acknowledgments
We thank Maxime Schwartz and Lluis Quintana-Murci for many helpful discussions and Xavier Nouvel for help with the manuscript.
This work received support from the European Commission (grant QLK2-CT-2000-00630), and from Louis D. French Academy of Sciences award.
References
- Raviglione MC, Gupta R, Dye CM, Espinal MA. The burden of drug-resistant tuberculosis and mechanisms for its control. Ann N Y Acad Sci. 2001;953:88–97. DOIPubMedGoogle Scholar
- Farmer P, Kim JY. Community based approaches to the control of multidrug resistant tuberculosis: introducing “DOTS-plus.”. BMJ. 1998;317:671–4.PubMedGoogle Scholar
- Telzak EE, Sepkowitz K, Alpert P, Mannheimer S, Medard F, el-Sadr W, Multidrug-resistant tuberculosis in patients without HIV infection. N Engl J Med. 1995;333:907–11. DOIPubMedGoogle Scholar
- Bifani PJ, Mathema B, Liu Z, Moghazeh SL, Shopsin B, Tempalski B, Identification of a W variant outbreak of Mycobacterium tuberculosis via population-based molecular epidemiology. JAMA. 1999;282:2321–7. DOIPubMedGoogle Scholar
- Bifani PJ, Mathema B, Kurepina NE, Kreiswirth BN. Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains. Trends Microbiol. 2002;10:45–52. DOIPubMedGoogle Scholar
- Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PW, Martin C, Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol. 1999;37:2607–18.PubMedGoogle Scholar
- Glynn JR, Whiteley J, Bifani PJ, Kremer K, van Soolingen D. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis. 2002;8:843–9.PubMedGoogle Scholar
- Anh DD, Borgdorff MW, Van LN, Lan NT, van Gorkom T, Kremer K, Mycobacterium tuberculosis Beijing genotype emerging in Vietnam. Emerg Infect Dis. 2000;6:302–5. DOIPubMedGoogle Scholar
- Bifani PJ, Plikaytis BB, Kapur V, Stockbauer K, Pan X, Lutfey ML, Origin and interstate spread of a New York City multidrug-resistant Mycobacterium tuberculosis clone family. JAMA. 1996;275:452–7. DOIPubMedGoogle Scholar
- Caminero JA, Pena MJ, Campos-Herrero MI, Rodriguez JC, Garcia I, Cabrera P, Epidemiological evidence of the spread of a Mycobacterium tuberculosis strain of the Beijing genotype on Gran Canaria Island. Am J Respir Crit Care Med. 2001;164:1165–70.PubMedGoogle Scholar
- Pfyffer GE, Strassle A, van Gorkum T, Portaels F, Rigouts L, Mathieu C, Multidrug-resistant tuberculosis in prison inmates, Azerbaijan. Emerg Infect Dis. 2001;7:855–61. DOIPubMedGoogle Scholar
- Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 1998;79:3–29. DOIPubMedGoogle Scholar
- Van Rie A. Analysis for a limited number of gene codons can predict drug resistance of Mycobacterium tuberculosis in a high incidence community. J Clin Microbiol. 2001;39:636–41. DOIPubMedGoogle Scholar
- Telenti A, Imboden P, Marchesi F, Lowrie D, Cole S, Colston MJ, Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet. 1993;341:647–50. DOIPubMedGoogle Scholar
- Finken M, Kirschner P, Meier A, Wrede A, Bottger EC. Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol Microbiol. 1993;9:1239–46. DOIPubMedGoogle Scholar
- Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–4. DOIPubMedGoogle Scholar
- Giraud A, Matic I, Tenaillon O, Clara A, Radman M, Fons M, Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science. 2001;291:2606–8. DOIPubMedGoogle Scholar
- Richardson AR, Stojiljkovic I. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol Microbiol. 2001;40:645–55. DOIPubMedGoogle Scholar
- Horst JP, Wu TH, Marinus MG. Escherichia coli mutator genes. Trends Microbiol. 1999;7:29–36. DOIPubMedGoogle Scholar
- Mizrahi V, Andersen SJ. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol Microbiol. 1998;29:1331–9. DOIPubMedGoogle Scholar
- David HL, Newman CM. Some observations on the genetics of isoniazid resistance in the tubercle bacilli. Am Rev Respir Dis. 1971;104:508–15.PubMedGoogle Scholar
- Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol. 2001;39:3563–71. DOIPubMedGoogle Scholar
- Kurepina N, Ramaswamy S, Shashkina EF, Sloutsky AM, Blinova LN, Mishustin SP, The sequence analysis of the pncA gene determining the PZA-resistance in the predominant M. tuberculosis strains isolated in the Tomsk penitentiary system, Western Siberia, Russia. In: 32nd World Conference on Lung Health of the International Union against Tuberculosis and Lung Disease. Paris, France; 2001.
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;94:9869–74. DOIPubMedGoogle Scholar
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. DOIPubMedGoogle Scholar
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. DOIPubMedGoogle Scholar
- Fujii Y, Shimokawa H, Sekiguchi M, Nakabeppu Y. Functional significance of the conserved residues for the 23-residue module among MTH1 and MutT family proteins. J Biol Chem. 1999;274:38251–9. DOIPubMedGoogle Scholar
- Maki H, Sekiguchi M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature. 1992;355:273–5. DOIPubMedGoogle Scholar
- Margison GP, Cooper DP, Potter PM. The E. coli ogt gene. Mutat Res. 1990;233:15–21. DOIPubMedGoogle Scholar
- Boiteux S, O’Connor TR, Laval J. Formamidopyrimidine-DNA glycosylase of Escherichia coli: cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J. 1987;6:3177–83.PubMedGoogle Scholar
- Au KG, Clark S, Miller JH, Modrich P. Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc Natl Acad Sci U S A. 1989;86:8877–81. DOIPubMedGoogle Scholar
- Andersson GE, Sharp PM. Codon usage in the Mycobacterium tuberculosis complex. Microbiology. 1996;142:915–25. DOIPubMedGoogle Scholar
- Kruuner A, Hoffner SE, Sillastu H, Danilovits M, Levina K, Svenson SB, Spread of drug-resistant pulmonary tuberculosis in Estonia. J Clin Microbiol. 2001;39:3339–45. DOIPubMedGoogle Scholar
- Tenaillon O, Toupance B, Le Nagard H, Taddei F, Godelle B. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics. 1999;152:485–93.PubMedGoogle Scholar
- Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700–2. DOIPubMedGoogle Scholar
- Rainey BP, Moxon ER. Microbiology. When being hyper keeps you fit. Science. 2000;288:1186–7. DOIPubMedGoogle Scholar
- Werngren J, Hoffner SE. Drug-susceptible Mycobacterium tuberculosis Beijing genotypes does not develop mutation-conferred resistance to rifampin at an elevated rate. J Clin Microbiol. 2003. In press. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 9, Number 7—July 2003
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Brigitte Gicquel, Unité de Génétique Mycobactérienne, Institut Pasteur, 28 rue du Dr. Roux, 75015 Paris, France; fax: 33145688843
Top