Volume 11, Number 8—August 2005
Dispatch
West Nile Virus Detection in Urine
Figure
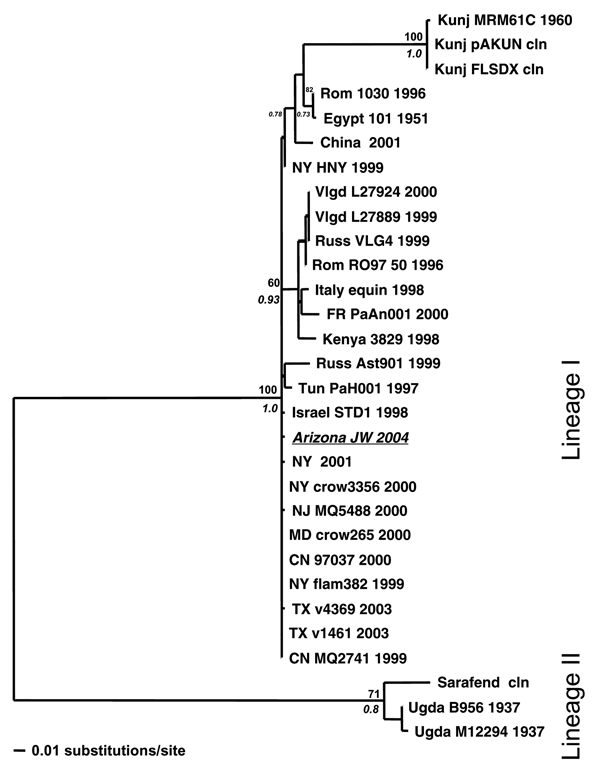
Figure. . Maximum likelihood (ML) tree showing the phylogenetic relationships between West Nile virus (WNV) urine sample Arizona JW 2004 (italicized and underlined) and previously published WNV strains based on capsid/prM gene junction (356 bp). Samples are coded by location, strain, and year of isolation. Locations include France (FR); Kunjin (Kunj); Romania (Rom); Russia (Russ); Tunisia (Tun); Uganda (Ugda); Volgograd, Russia (Vlgd); and the US states of New York (NY), Texas (TX), New Jersey (NJ), Maryland (MD), and Connecticut (CN). Support indicated above and below nodes are bootstrap values (1,000 neighbor-joining replicates using the ML model of evolution) and Bayesian posterior probabilities (Bayesian MCMC [Metropolis-Hastings Markov chain Monte Carlo tree sampling] for 4 chains length 2 × 106, sample frequency 100, with a 2,000-tree burn-in), respectively.