Volume 16, Number 6—June 2010
Dispatch
Novel Betaherpesvirus in Bats
Cite This Article
Citation for Media
Abstract
Because bats are associated with emerging zoonoses, identification and characterization of novel viruses from bats is needed. Using a modified rapid determination system for viral RNA/DNA sequences, we identified a novel bat betaherpesvirus 2 not detected by herpesvirus consensus PCR. This modified system is useful for detecting unknown viruses.
Since the 1990s, bats have been associated with several emerging zoonotic agents, including Hendra, Nipah, Ebola, lyssa, and severe acute respiratory syndrome coronavirus-like viruses (1). Bats seem to have great potential as reservoirs for emerging viruses. Therefore, to understand the role of bats as a host species, identification and characterization of novel viruses from bats is needed. For virus isolation, we have been attempting to establish primary cell cultures from various bats (2,3). Using a rapid determination system for viral RNA sequences (RDV), we discovered a novel adenovirus and gammaherpesvirus in bats (2,4). This system, which we simplified to a less laborious one (5), is useful for detecting viruses, regardless of virus species (6).
During June–August, 2008, with the permission of the governor of Wakayama Prefecture, Japan, we caught 8 insectivorous vespertilionid bats, Miniopterus fuliginosus, and used their spleens and kidneys to establish primary cell cultures. During passage of the primary spleen adherent cells, cytopathic effect (cell death) was noted at third passage. The collected supernatant was injected into fresh primary kidney cells and caused apparent cytopathic effect at first passage.
Before using the RDV method, we had attempted to detect herpesvirus by nested PCR with the consensus primer sets DFA, ILK, KG1, TGV, and IYG, which were designed according to the consensus-degenerate hybrid oligonucleotide primers program (7). These consensus degenerate primers are effective for detecting many herpesviruses from vertebrate hosts. However, in this study they failed to detect any herpesviruses.
We then attempted to detect herpesvirus by using RDV version 3.1, our modification from version 3.0 (5). The adapters and primers for construction of the second cDNA library in RDV version 3.1 were newly designed and replaced those used in RDV version 3.0 (Technical Appendix 1). Both adapters have sticky-end structures digested with Sau3AI or HpyCH4 IV. RDV version 3.1 can determine an unknown viral cDNA fragment with 64 primer pairs, which we used for constructing the second cDNA library.
Appendix Figure
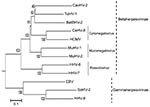
Appendix Figure. Phylogenetic tree based on the deduced amino acid sequences of complete glycoprotein B. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates)...
With RDV version 3.1, we obtained 4 unknown cDNA fragments, which had no matches in a BLASTn (www.ncbi.nlm.nih.gov/blast/Blast.cgi) search. In a BLASTx search, 1 cDNA fragment (deduced sequence of 29 aa) was homologous to the glycoprotein B (gB) amino acid sequence of the tupaiid herpesvirus 1 (TuHV-1) (79% identity), which belongs to subfamily Betaherpesvirinae. We designed new consensus-degenerate hybrid oligonucleotide primers (http://blocks.fhcrc.org/codehop.html) selective for the betaherpesvirus gB and DNA polymerase (DPOL) genes, and we determined the complete gB sequence and the partial DPOL sequence of the isolated virus (5,029 bp, DNA Data Bank of Japan accession no. AB517983). BLAST search indicated that the complete gB sequence was novel and most similar to that of TuHV-1 (59% aa sequence identity) (Appendix Figure). We named the isolated virus bat betaherpesvirus 2 (BatBHV-2).
Figure
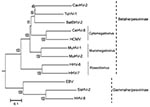
Figure. Phylogenetic tree based on the deduced amino acid sequences of complete glycoprotein B. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates)...
We constructed a phylogenetic tree by using the neighbor-joining method with the gB amino acid sequence and the available sequences of known herpesviruses (Figure). The phylogenetic tree based on betaherpesvirus gB genes showed that BatBHV-2 is most closely related to TuHV-1 and caviid herpesvirus 2 (guinea pig cytomegalovirus). The subfamily Betaherpesvirinae consists of the genera Cytomegalovirus, Muromegalovirus, and Roseolovirus. TuHV-2 and caviid herpesvirus 2 are species unassigned to any genus in the subfamily Betaherpesvirinae.
In May 2009, we collected, again with permission, another 50 bats belonging to 1 species, M. fuliginosus, from the same location for an epizootologic study (Technical Appendix 2). Spleens and blood were collected from all bats, and other organs (liver, kidney, lung, brain, intestine, trachea, and urinary bladder) were collected from 10 bats. Nested PCR was performed by using specific primers selective for the DPOL gene of BatBHV-2, and PCR products were subjected to direct sequencing. Viral nucleotide sequences were obtained from 4 of the 50 spleen samples. Each nucleotide sequence showed complete identity to the partial DPOL sequence of the BatBHV-2. Other organs and serum collected from 2 of the bats were also tested by nested PCR, and viral DNA was detected in the liver, kidneys, and lungs of both bats.
Although PCRs with consensus primers effectively detect known and unknown viruses, they failed to detect BatBHV-2, possibly because of minor mismatches between the sequences of BatBHV-2 and the primer sets (TGV, IYG, and KG1). The variety of virus sequences and gene mutations often prevents successful amplification of virus genes. RDV, however, can detect viral cDNA fragments independent of virus species and thus is useful as a first-choice tool for identifying emerging known and unknown viruses in animals and humans.
BLAST search showed that the complete gB sequence of the isolated virus was novel and most similar to that of TuHV-1. Recently, bats have been described as hosts for herpesviruses in several countries in Europe, America, Africa, and Asia (4,9,10). Wibbelt et al. reported that the partial DPOL sequence (175 bp) of a betaherpesvirus, bat betaherpesvirus 1 (BatBHV-1), was obtained from several insectivorous bat species (10). Although the length of the BatBHV-1 sequence was short, similarity between BatBHV-1 and BatBHV-2 was relatively high (58%). BatBHV-1 is most similar to TuHV-1(61%). These findings suggest that BatBHV-2 is a different species than BatBHV-1.
Our epizootologic study found relatively high (8%) prevalence of BatBHV-2 in insectivorous bats. Although the virus genome was detected in a few parenchymal organs by nested PCR, no amplification was possible for serum, intestine, or urinary bladder samples, which may exclude apparent virus shedding by the bats. In addition, all 50 bats collected appeared clinically healthy. To understand the life cycle of this virus, the possibility of a latent infection in these insectivorous bats must be explored.
Mr Watanabe is a PhD student in the Department of Veterinary Microbiology, Graduate School of Agricultural and Life Sciences, The University of Tokyo. His research interests include the epidemiology and pathogenic mechanisms of emerging viruses from bats.
Acknowledgments
We thank Momoko Ogata and Miho Nishimura for their assistance.
This study was supported in part by grants from the Japan Society for the Promotion of Science; the Ministry of Health, Labor, and Welfare; and the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
References
- Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev Med Virol. 2007;17:67–91. DOIPubMedGoogle Scholar
- Maeda K, Hondo E, Terakawa J, Kiso Y, Nakaichi N, Endoh D, Isolation of novel adenovirus from fruit bat (Pteropus dasymallus yayeyamae). Emerg Infect Dis. 2008;14:347–9. DOIPubMedGoogle Scholar
- Omatsu T, Watanabe S, Akashi H, Yoshikawa Y. Biological characters of bats in relation to natural reservoir of emerging viruses. Comp Immunol Microbiol Infect Dis. 2007;30:357–74. DOIPubMedGoogle Scholar
- Watanabe S, Ueda N, Iha K, Masangkay JS, Fujii H, Alviola P, Detection of a new bat gammaherpesvirus in the Philippines. Virus Genes. 2009;39:90–3. DOIPubMedGoogle Scholar
- Watanabe S, Mizutani T, Sakai K, Kato K, Tohya Y, Fukushi S, Ligation-mediated amplification for effective rapid determination of viral RNA sequences (RDV). J Clin Virol. 2008;43:56–9. DOIPubMedGoogle Scholar
- Mizutani T, Endoh D, Okamoto M, Shirato K, Shimizu H, Arita M, Rapid genome sequencing of RNA viruses. Emerg Infect Dis. 2007;13:322–4. DOIPubMedGoogle Scholar
- VanDevanter DR, Warrener P, Bennett L, Schultz ER, Coulter S, Garber RL, Detection and analysis of diverse herpesviral species by consensus primer PCR. J Clin Microbiol. 1996;34:1666–71.PubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Razafindratsimandresy R, Jeanmaire EM, Counor D, Vasconcelos PF, Sall AA, Reynes JM. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. J Gen Virol. 2009;90:44–7. DOIPubMedGoogle Scholar
- Wibbelt G, Kurth A, Yasmum N, Bannert M, Nagel S, Nitsche A, Discovery of herpesviruses in bats. J Gen Virol. 2007;88:2651–5. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 16, Number 6—June 2010
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Tetsuya Mizutani, Department of Virology 1, National Institute of Infectious Diseases, Gakuen 4-7-1, Musashimurayama, Tokyo 208-0011, Japan
Top