Volume 17, Number 2—February 2011
Dispatch
Usefulness of Published PCR Primers in Detecting Human Rhinovirus Infection
Cite This Article
Citation for Media
Abstract
We conducted a preliminary comparison of the relative sensitivity of a cross-section of published human rhinovirus (HRV)–specific PCR primer pairs, varying the oligonucleotides and annealing temperature. None of the pairs could detect all HRVs in 2 panels of genotyped clinical specimens; >1 PCR is required for accurate description of HRV epidemiology.
Human rhinoviruses (HRVs) cause more asthma exacerbations than any other known factor, in addition to causing most colds and influenza-like illnesses. The prevalence of HRV in published reports varies considerably. A novel HRV clade identified in 2006, now known as HRV species C (HRV-C) (1), can be identified only by PCR. Since 1988, seasonality and clinical outcomes and numerous different primer pairs have been used to identify HRV; how well these methods perform on new HRV types is uncertain. Given the likely variation in the preparation of RNA, the quality and formulations of commercial reverse transcription (RT)-PCR enzymes and reaction mix components and changes in thermal cyclers since 1988, not surprisingly many, perhaps most, of these assays are not being used in the manner they were originally described. For example, the first HRV-specific primers reported (2) have subsequently been used with different RNA preparation methods, amounts of reverse transcriptase, cDNA priming strategies, dNTP concentrations, annealing temperatures (TMs), and cycling conditions (3,4).
Figure
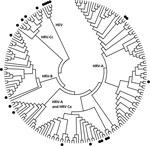
Figure. Distribution of human rhinovirus (HRV) and human enterovirus (HEV) sequences used for primer pair studies. The HRV and HEV genotypes from the testing panel (indicated by filled circles) were aligned with...
We conducted a preliminary comparison of the relative sensitivity of a cross-section of published HRV-specific PCR primer pairs (most of which were first published before HRV-C was reported), independent of most variables described above, by testing a panel of 57 clinical specimen nucleic acid extracts from combined nose and throat swabs from preschool children with colds and influenza-like illnesses in Melbourne, Australia. The study was approved by the Royal Children’s Hospital Human Research Ethics Committee. The panel included representatives of the 3 HRV species (Figure), human enteroviruses (HEVs), and extracts negative for picornaviruses. The HRVs had been previously detected by using a nested primer pair (Table A1) (5). We used 10 different HRV primer pairs and also retested specimens by using the original primer pair with our standard reagents and equipment (5). We applied the published TM when possible. The original descriptions of primer pairs 7 and 10 (Table A1) lacked TM information, and after in-house calculations, we used TMs of 50°C and 58°C, respectively. We also deliberately standardized the reagents (OneStep RT-PCR kit, QIAGEN, Doncaster, Victoria, Australia) and thermal cyclers used (Veriti, Applied Biosystems, Foster City, CA, USA) for conventional PCR and the RotorGene 3000 real-time cycler (QIAGEN). Because primer pair 1 had a published history of detecting types from all HRV species, we chose it to genotype HRV-positive samples by sequencing the amplified products. Other pairs were used if pair 1 was unsuccessful.
We found that no primer pair detected the same HRVs and HEVs typed when the original pair (5) or pair 1 (Table A1) was used. Five primer pairs, including real-time PCR (rtPCR) pair 5, did not amplify the HEVs, a positive feature for HRV-specific studies. Only 2 primer pairs amplified anything from a specimen that was positive for both HRV and HEV, a problem for accurate estimation of the frequency of co-detections. The original primer pair screen detected 3 untypeable picornaviruses, which were not detected by any other pair or by repeat testing using the same pair. Only the second-round amplicon of the 3 nested sets of nested primer pairs (2,3, and,9) was considered because the second round increased the total number of positive specimens over the first round. The longest amplicon, produced by primer pair 7, was also a valuable genotyping target, but it detected only 14 of the original 27 HRV-positive specimens in this population.
We next selected 4 frequently published primer pairs (1,5,7, and,8) to examine 44 picornavirus-positive specimens (39 HRVs, 3 HEVs, and 2 untypeable picornaviruses) from nonhospitalized children with acute asthma exacerbation (6). As before, primer pair 1 detected the greatest number of HRV- and HEV- positive specimens and all positive specimens detected by other primer sets (n = 41), followed by pair 7 (n = 40), pair 5 (n = 36), and pair 8 (n = 31). Most notably, primer pair 7 performed better than it had in the previous population, detecting only 1 fewer HRV than primer pair 1 and 9 more HRVs than pair 8. No species-specific bias was apparent, but generally, a specimen with a lower RNA concentration, as indicated by the cycle threshold from primer pair 5, was less likely to be detected or typed by using other primer pairs. Primer pairs 5 and 8 did not detect the 3 HEVs (HEV-68). We noted in both populations that primer pair 1 sometimes amplified a region of human genomic DNA from chromosome 6 (GQ497714), for which amplicon size was indistinguishable from that expected due to HRV.
It was not possible to use the precise conditions reported for the 10 compared assays; 1 was published >2 decades ago and used phenol chloroform extraction. Some of the original enzyme formulations or reagents are no longer available, and production processes have changed in the interim. Thermal cyclers have also changed. There was no consensus on enzymes and reaction mixes used. In addition, the previously published primers were used in assays divided between those using 1-step RT-PCR and those using a separate RT cDNA synthesis step. A review of studies that detected HRVs with adequately described conditions during 2009–2010 found that fewer used a single-tube RT-PCR approach than a 2-step system. We conducted single-tube RT-PCR to maintain the benefits of the so-called closed amplification system of rtPCR. Thus, we chose to use a single common set of reagents as the fairest way to compare the primer pairs examined in this study. We believe the nature of this relative comparison best reflects performance for the likely end users: clinical microbiology laboratories or researchers.
We compared primers rather than assay function using clinical material instead of cultured virus, plasmid or synthetic RNA standards, or screening contemporary or archived extracts, which are sometimes of low viral load. When picornavirus epidemiology is the primary research focus, we recommend using >2 primer pairs to maximize the detection of HRVs. Under our conditions, pairs 1–4 returned the highest number of positive results, and the rtPCRs behaved similarly but with reduced sensitivity. The rtPCR that used pair 5 did not amplify known HEVs.
Many possible reasons could cause discrepant virus testing results between different sites, including changes to specimen integrity resulting from transport and variable amplification resulting from low viral loads. The effects of viral load can be seen in this study: specimens in population 1 that were positive with multiple (>6 separate pairs) primer pairs had a mean cycle threshold of 33.3 (combining results from both rtPCRs), whereas those with <6 positive results had means of 39.3 cycles. Most (29/33) specimens with <3 positive primer pairs were negative by rtPCR. Amplification variability can also be attributed to the substantial nucleotide sequence diversity between HRVs and the different temporal and clinical characteristics of the 2 specimen populations we used. Population diversity is a feature of HRV studies in the literature.
Our selection of published primer pairs includes those from studies that have informed our current understanding of HRV epidemiology. Finding such a high degree of variability in performance was thus noteworthy. Inefficient HRV detection by PCR may be a serious problem for research studies. Comparison of data between different HRV studies is confounded as are data from studies seeking to determine the effects of other respiratory viruses. The prevalence, seasonality, transmission, and clinical effects of HRV types and species require reexamination with tools that have been comparatively validated to ensure their sensitivity.
Ms Faux conducted this study as a research scientist in the Queensland Paediatric Infectious Diseases Laboratory, Sir Albert Sakzewski Virus Research Centre. Her main field of research was the detection and characterization of newly identified and classic respiratory viruses in children.
Acknowledgment
This study was supported by the National Health and Medical Research Council, Australia, Project Grant 455905, and Queensland Children’s Medical Research Institute Research Project Seeding Grant (Established Researcher) 10281.
References
- Arden KE, McErlean P, Nissen MD, Sloots TP, Mackay IM. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J Med Virol. 2006;78:1232–40.DOIPubMedGoogle Scholar
- Gama RE, Horsnell PR, Hughes PJ, North C, Bruce CB, Al-Nakib W, Amplification of rhinovirus specific nucleic acids from clinical samples using the polymerase chain reaction. J Med Virol. 1989;28:73–7.DOIPubMedGoogle Scholar
- Papadopoulos NG, Sanderson G, Hunter J, Johnston SL. Rhinoviruses replicate effectively at lower airway temperatures. J Med Virol. 1999;58:100–4.DOIPubMedGoogle Scholar
- Winther B, Hayden FG, Hendley JO. Picornavirus infections in children diagnosed by RT-PCR during longitudinal surveillance with weekly sampling: association with symptomatic illness and effect of season. J Med Virol. 2006;78:644–50.DOIPubMedGoogle Scholar
- Lambert SB, Allen KM, Druce JD, Birch CJ, Mackay IM, Carlin JB, Community epidemiology of human metapneumovirus, human coronavirus NL63, and other respiratory viruses in healthy preschool-aged children using parent-collected specimens. Pediatrics. 2007;120:e929–37.DOIPubMedGoogle Scholar
- Arden KE, Chang AB, Lambert SB, Nissen MD, Sloots TP, Mackay IM. Newly identified respiratory viruses in children with non-hospitalised asthma exacerbation. J Med Virol. 2010;82:1458–61.DOIPubMedGoogle Scholar
- Gama RE, Hughes PJ, Bruce CB, Stanway G. Polymerase chain reaction amplification of rhinovirus nucleic acids from clinical material. Nucleic Acids Res. 1988;16:9346.DOIPubMedGoogle Scholar
- Steininger C, Aberle SW, Popow-Kraupp T. Early detection of acute rhinovirus infections by a rapid reverse transcription–PCR assay. J Clin Microbiol. 2001;39:129–33.DOIPubMedGoogle Scholar
- Kiang D, Yagi S, Kantardjieff KA, Kim EJ, Louie JK, Schnurr DP. Molecular characterization of a variant rhinovirus from an outbreak associated with uncommonly high mortality. J Clin Virol. 2007;38:227–37.DOIPubMedGoogle Scholar
- Lu X, Holloway B, Dare RK, Kuypers J, Yagi S, Williams JV, Real-time reverse transcription–PCR assay for comprehensive detection of human rhinoviruses. J Clin Microbiol. 2008;46:533–9.DOIPubMedGoogle Scholar
- Tapparel C, Junier T, Gerlach D, Van-Belle S, Turin L, Cordey S, New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerg Infect Dis. 2009;15:719–26.PubMedGoogle Scholar
- Savolainen C, Blomqvist S, Mulders MN, Hovi T. Genetic clustering of all 102 human rhinovirus prototype strains: serotype 87 is close to human enterovirus 70. J Gen Virol. 2002;83:333–40.PubMedGoogle Scholar
- Arola A, Santti J, Ruuskanen O, Halonen P, Hyypiä T. Identification of enteroviruses in clinical specimens by competitive PCR followed by genetic typing using sequence analysis. J Clin Microbiol. 1996;34:313–8.PubMedGoogle Scholar
- Coiras MT, Aguilar JC, García ML, Casas I, Pérez-Breña MP. Simultaneous detection of fourteen respiratory viruses in clinical specimens by two multiplex reverse transcription nested-PCR assays. J Med Virol. 2004;72:484–95.DOIPubMedGoogle Scholar
- Gunson RN, Collins TC, Carman WF. Real-time RT-PCR detection of 12 respiratory viral infections in four triplex reactions. J Clin Virol. 2005;33:341–4.DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleTable of Contents – Volume 17, Number 2—February 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Ian M. Mackay, Queensland Children’s Medical Research Institute, Royal Children’s Hospital–Queensland Paediatric Infectious Diseases Laboratory, Sir Albert Sakzewski Virus Research Centre, Brisbane, Queensland, Australia
Top