Volume 18, Number 2—February 2012
Letter
Recombination of Human Coxsackievirus B5 in Hand, Foot, and Mouth Disease Patients, China
Cite This Article
Citation for Media
To the Editor: Hand, foot, and mouth disease (HFMD) is an acute viral infectious disease in infants and young children. However, since 2008, HFMD has emerged as a major public health problem in the People’s Republic of China, resulting in millions of infections with hundreds of deaths (1). Human enteroviruses (HEVs), including HEV71, echoviruses, and coxsackie viruses A and B (CAV and CBV), are the major pathogens of HFMD (2). In mainland China, HEV71 and CAV16 have been recognized as the dominant causative agents for HFMD.
Figure
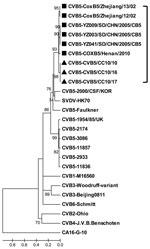
Figure. . Phylogenetic analysis of selected human coxsackievirus B (CBV) strains from different origins based on the viral protein 1 gene sequences. The neighbor-joining tree was generated by using MEGA4 software (<>
During a recent HFMD outbreak in Changchun during 2010, three of 16 throat swab samples tested positive for HEV but negative for HEV71 and CAV16 by reverse transcription PCR. All 3 samples were then placed into human rhabdomyosarcoma cells, and typical cytopathic effects were observed 3–4 days later. All the isolates were finally characterized as CBV5 by using serologic and molecular technology and designated as CBV5/CC10/10, CBV5/CC10/16, and CBV5/CC10/17, respectively. The complete genome of these Changchun isolates was determined as described (3) and submitted to GenBank (accession nos. JN580070, JN695050, and JN695051, respectively. The genome RNA of CBV5/CC10/10 is 7,402 bp long, and the 5′ and 3′ untranslated regions are 743 and 101 bp, respectively. The coding regions of these Changchun isolates are highly homologous, with amino acid identity of 100% and only a 3-nt difference exists among them. The complete genome of 4 CBV5 strains were indexed previously in GenBank, and the nucleotide sequence identities of the newly isolated CBV5/CC10/10 with the other 4 CBV5 strains were among 80.6%–88.1%. Results of homology and phylogenetic analyses based on the complete viral protein 1 sequence (849 bp) showed that the nucleotide identity of viral protein 1 among 17 different CBV5 strains was 78.9%–95.6% and the amino acid identity was 92.9%–98.9%. The neighbor-joining tree indicated that the new isolated CBV5 Changchun strains were most closely related to the strains isolated from mainland China and that they divided into a distinct lineage from other CBV5 strains outside China (Figure). CBV5 infections were reported in mainland China during 2002–2010 in Zhejiang, Shandong, and Henan Provinces (4). These Changchun isolates were highly homologous with the recent Henan isolate, COXB5/Henan/2010, with a nucleotide identity of 88.1%. These results indicated that CBV5 might have been circulating in China for many years and represented an independent evolution tendency.
Homology and BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) based on the complete genome sequence showed that these newly CBV5 isolates have 85% identity with some human CBV3 strains. Because RNA recombination is a well-known phenomenon for HEVs during viral evolution and reemergence (5–8), recombination analysis between newly isolated CBV5 and other HEVs was performed by using SimPlot software. Similarity scanning analysis (Technical Appendix Figure 1) by using CBV5/CC10/10 as query sequence showed that the 5′ half (nt 1–4481) of the genome had high similarity (>93%) to CBV5 strain COXB5/Henan/2010, and the 3′ half (nt 4661–7402) showed high similarity (>97%) to CBV3 strain Beijing0811. Then, bootscanning analysis (Technical Appendix Figure 2) showed that CBV5/CC10/10 was most closely related to COXB5/Henan/2010 in the 5′ half of the genome, and to CBV3 Beijing0811 in the 3′ half of the genome. Both analyses showed that the small fragment within 2C domains are highly similar (>84%) to a CBV5 strain from South Korea, CBV5/2000/CSF/KOR. Genetic algorithms for recombination detection analysis also indicated that the putative breakpoints were located within the 2C domain. We found no recombinant evidence between CBV5 and other HEVs during the analysis. Together, these accordant results demonstrated that recombination has possibly occurred within the 2C domain, and these Changchun isolates are possibly progeny of intertypic and intratypic recombination of CBV strains circulating in China (COXB5/Henan/2010 and Beijing0811) and South Korea (2000/CSF/KOR).
HEVs can be divided into ≈100 serotypes, and intertypic or intratypic recombination among different viruses occurs frequently, which is information necessary for disease control and surveillance. In China, dozens of HEVs, including HEV71, CAV, CBV, and echoviruses, have been isolated from HFMD patients and identified as the pathogens causing HFMD (1,2,9,10). Recombination events among HEVs have been shown to play roles in HFMD outbreaks. A recombinant HEV71 with CAV16 was suggested to be responsible for the HFMD outbreak in Fuyang, China, in 2008 (9). Recombination also occurred among the CAV (types 2, 4, 5, and 10) isolates during a HFMD outbreak in China during 2009 (10). However, most recombinant studies have focused on CAV and poliovirus. Recently, Oberste et al. (6) proposed the possible role of recombination within CBV strains based on phylogeneic analysis of individual genes. Our current study identified and characterized the newly CBV5 isolates from HFMD patient as intratypic and intertypic recombinants, and further surveillance is warranted that focuses on the emerging recombinant viruses among HFMD causative agents and investigations on the pathogenic role and disease associations of the recombinant CBVs.
Acknowledgment
This study was supported in part by the Major Special Program of National Science and Technology of China (no.2009ZX10004-204), the National Natural Science Foundation of China (no. 81000721), and Beijing Natural Science Foundation (no. 7112108). C.F.Q. was supported by Beijing Nova Program of Science and Technology (no.2010B041).
References
- Qiu J. Viral outbreak in China tests government efforts. Nature. 2009;458:554–5. DOIPubMedGoogle Scholar
- Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010;10:778–90. DOIPubMedGoogle Scholar
- Lindberg AM, Polacek C. Molecular analysis of the prototype coxsackievirus B5 genome. Arch Virol. 2000;145:205–21. DOIPubMedGoogle Scholar
- Wang HY, Li Y, Xu AQ, Zhang Y, Tao ZX, Liu GF, Identification and phylogenic analysis of Coxsackie-virus B5 during an outbreak of aseptic meningitis in Shandong [in Chinese]. Zhonghua Liu Xing Bing Xue Za Zhi. 2010;31:64–8.PubMedGoogle Scholar
- Lukashev AN. Role of recombination in evolution of enteroviruses. Rev Med Virol. 2005;15:157–67. DOIPubMedGoogle Scholar
- Oberste MS, Penaranda S, Pallansch MA. RNA recombination plays a major role in genomic change during circulation of coxsackie B viruses. J Virol. 2004;78:2948–55. DOIPubMedGoogle Scholar
- Simmonds P, Welch J. Frequency and dynamics of recombination within different species of human enteroviruses. J Virol. 2006;80:483–93. DOIPubMedGoogle Scholar
- Zhao K, Han X, Wang G, Hu W, Zhang W, Yu XF. Circulating coxsackievirus A16 identified as recombinant type A human enterovirus, China. Emerg Infect Dis. 2011;17:1537–40.PubMedGoogle Scholar
- Zhang Y, Zhu Z, Yang W, Ren J, Tan X, Wang Y, An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of hand foot and mouth disease in Fuyang city of China. Virol J. 2010;7:94. DOIPubMedGoogle Scholar
- Hu YF, Yang F, Du J, Dong J, Zhang T, Wu ZQ, Complete genome analysis of coxsackievirus A2, A4, A5, and A10 strains isolated from hand, foot, and mouth disease patients in China revealing frequent recombination of human enterovirus A. J Clin Microbiol. 2011;49:2426–34. DOIPubMedGoogle Scholar
Figure
Cite This ArticleRelated Links
Table of Contents – Volume 18, Number 2—February 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Cheng-Feng Qin, Department of Virology, State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Beijing 100071, People’s Republic of China
Top