Volume 18, Number 4—April 2012
Dispatch
Characterization of Mycobacterium orygis as M. tuberculosis Complex Subspecies
Cite This Article
Citation for Media
Abstract
The oryx bacilli are Mycobacterium tuberculosis complex organisms for which phylogenetic position and host range are unsettled. We characterized 22 isolates by molecular methods and propose elevation to subspecies status as M. orygis. M. orygis is a causative agent of tuberculosis in animals and humans from Africa and South Asia.
Traditionally, the Mycobacterium tuberculosis complex comprises tubercle bacilli of 8 distinct subgroups: M. tuberculosis, M. africanum, M. canettii, M. bovis, M. caprae, M. pinnipedii, M. microti, and M. mungi (1–4). Two other distinct branches of the M. tuberculosis complex phylogenetic tree exist, the dassie and oryx bacilli, causative agents of tuberculosis in the animal species after which they are named. Neither has been validly described as separate taxa, nor have they been associated with disease in humans (1–4).
Oryx bacilli have been isolated from members of the Bovidae family, i.e., oryxes, gazelles (3), deer, antelope, and waterbucks (5), although their exact host range remains unsettled. No human disease caused by the oryx bacilli has been reported. These bacilli most likely constitute a separate phylogenetic lineage; however, their exact position has not been established with valid phylogenetic markers, such as large genomic deletions or single nucleotide polymorphisms (SNPs). To settle the phylogenetic position and host range of the oryx bacilli, we collected all oryx bacillus isolates from our laboratory database to establish their sources and subjected the isolates to extended phylogenetic analysis.
Figure 1

Figure 1. Spoligotyping and 24-locus variable number tandem repeat (VNTR) typing results for Mycobacterium orygis. A) Spoligotyping patterns for the oryx bacillus isolates in this study; ST587 is the most common pattern (labeled...
We selected 22 isolates on the basis of >90% similarity of the IS6110 restriction fragment-length polymorphism (RFLP) pattern to that of established and previously published oryx bacillus strains; 11 isolates originated from animals, and 11 originated from 10 human patients (Figure 1) (1–3). All isolates yielded smooth to greasy domed nonchromogenic colonies in culture (Technical Appendix Figure).
For phylogenetic analysis, we performed SNP and region of difference (RD) analysis (2,6). RD and SNP typing showed a consistent pattern among the isolates, with presence of regions RD1, RD2, RD4, RD5a (Rv2348), RD6, and RD13–RD16 and absence of regions RD3, RD5b (plcA), and RD7–RD12 (Technical Appendix Table 1). The deleted region for RD12 (RD12oryx) was larger than that for M. bovis and M. caprae. Analysis of the flanking regions indicated an IS6110 insertion at the M. tuberculosis H37Rv coordinates of 3479670 and 3491252 with deletion of the intermediate area covering the open reading frames of the Rv3111 to Rv3125c genes (Technical Appendix Table 2). Isolates also showed the RDoryx_1, RDoryx_4, and RDoryx_wag22 deletions and the mmpL6551AAG mutation (Technical Appendix Table 1). Results agreed with those from previous studies (1,6).
Using pncA-1F 5′-GGC CGC GAT GAC ACC TCT-3′, pncA1-R 5′-GCC GCA GCC AAT TCA GCA GT-3′, pncA-2F 5′-CGA AGC GGC GGA CTA CCA TCA CG-3′, and pncA-2R 5′-CCC CAC CTG CGG CTG CGA ACC-3′ primers, we partially sequenced Rv2042c, Rv2044c, and the full pncA gene. The pncA sequences of the isolates from animals and humans were identical to those of M. tuberculosis H37Rv; in codon 38 of the Rv2042c gene, directly upstream from pncA, a GTC to GGC (Ser→Ala) mutation was noted in all 22 isolates; the partial Rv2042c sequence is stored in GenBank (accession no. JF417976). To assess the specificity of the Rv204238 GGC mutation, we screened 2 isolates of all M. tuberculosis complex (sub)species and 2 isolates of all M. tuberculosis groupings, on the basis of >60% IS6110 similarity, for this mutation; we did not find it in any of the strains tested (data not shown).
We performed spoligotyping and 24-locus mycobacterial interspersed repetitive units–variable-number tandem repeat (MIRU-VNTR) typing, as described (7,8). Spoligotyping mostly showed the sequence type (ST) 587 pattern in the spolDB4 database and labeled M. africanum (9); minor variations in spoligotype were observed (Figure 1, panel A). All isolates had unique IS6110 RFLP patterns, although with >75% similarity; patterns were characterized by high (i.e., 17–20) numbers of IS6110 copies (data not shown). VNTR typing showed closely related patterns (Table A1). A minimum spanning tree showed the clonality of the M. orygis isolates (Figure 1, panel B). The GenoType MTBC assay (Hain Lifesciences, Nehren, Germany) identified all isolates as M. africanum.
Baseline clinical data of humans were extracted from the anonymized National Tuberculosis Register. Ethical approval was waived for this retrospective laboratory-based study. Nine of the 10 human patients were of South Asian origin; the other was of Southeast Asian origin (Figure 1); patients’ average age was 41 years (range: 0–69 years). Clinically, 6 patients had pulmonary tuberculosis, 3 had lymphadenitis, and 1 child had tuberculosis diagnosed by gastric fluid culture. All isolates were susceptible to all first-line antituberculosis drugs, including pyrazinamide, and hence the standard treatment regimen was started for all patients. Patients received treatment for an average of 9 months; no details about individual regimens were available. No bacteriologically proven relapses were noted. No information was available about contact-tracing studies.
Figure 2
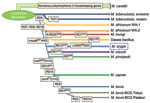
The oryx bacillus is a phylogenetically distinct lineage of the clonal M. tuberculosis complex and thus deserves a separate subspecies status; we propose the name M. orygis (Latin: oryx, genitive: orygis, of the oryx) to convey that this subspecies was first characterized after its isolation from an oryx (Figure 2).
The most common spoligotype (ST587) is present in the spolDB4 database and labeled M. africanum (9). The M. orygis bacteria share the gyrB1450 (G→T) mutation with M. africanum, M. microti, and M. pinnipedii (1). Hence, the GenoType MTBC assay identifies M. orygis as M. africanum. Thus, M. orygis isolates may have previously been misidentified as M. africanum (9,10).
The animal-adapted M. tuberculosis complex lineage is thought to have evolved in Africa when an M. africanum–like clone diverged from M. tuberculosis, as shown by the loss of the RD9 locus. Consecutive loss of DNA during the adaptation to novel hosts led to the distinct subspecies with its distinct host range that we know today (1,4,5,11). This matches geographically with the habitats of Oryx species, gazelles, and waterbucks.
For M. orygis, the host range remains unknown but may include oryxes, waterbucks, and gazelles in eastern Africa and the Arabian Peninsula; cows and rhesus monkeys in South Asia; and humans. The evolutionary explanation for the diversity in geographic distribution and hosts of M. orygis remains elusive. This diversity contrasts starkly with the conserved VNTR and spoligotype patterns.
The presence of M. orygis in diseased cows and a monkey in Bangladesh, unique RFLP patterns, and lack of onward transmission suggest animal-to-human transmission. As for M. bovis; humans may be accidental, dead-end hosts.
M. orygis, unlike M. microti, the dassie bacillus, and M. mungi, shows an intact RD1 region. This region encodes part of the virulence-related ESX-1 secretion system of tubercle bacilli (12).
Molecular characteristics define the isolates previously labeled as oryx bacilli as a distinct subspecies in the M. tuberculosis complex for which we propose the name M. orygis. The Rv204238 GGC mutation is a novel, useful genetic marker to identify M. orygis, which is otherwise characterized by the presence of genomic regions RD1, RD2, RD4, RD5a (Rv2348), RD6, RD13–RD16, and the mmpL6551AAG polymorphism, with absence of regions RD3, RD5b (plcA), RD7–RD12, RDoryx_1, RDoryx_4, and RDoryx_wag22. The deletion of RD12 is subspecies specific. Isolates yield the ST587 or closely related spoligotypes, 17–20 copies of IS6110, and a distinct 24-locus VNTR pattern with minor variations. M. orygis is a causative agent of tuberculosis in oryxes, gazelles, and waterbucks of African origin; cows and rhesus monkeys of South Asian origin; and humans.
Dr van Ingen is a resident in clinical microbiology at the Radboud University Nijmegen Medical Center. His primary research interests are the phylogeny and taxonomy of the genus Mycobacterium and treatment of tuberculosis and nontuberculous mycobacterial disease.
Acknowledgment
R.B. and R.S. were supported by the European Community’s Seventh Framework Program (FP7/2007-2013) under grant agreement no. 201762.
References
- Huard RC, Fabre M, de Haas P, Oliveira Lazzarini LC, van Soolingen D, Cousins D, Novel genetic polymorphisms that further delineate the phylogeny of the Mycobacterium tuberculosis complex. J Bacteriol. 2006;188:4271–87. DOIPubMedGoogle Scholar
- Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. DOIPubMedGoogle Scholar
- van Soolingen D, de Haas PEW, Haagsma J, Eger T, Hermans PWM, Ritacco V, Use of various genetic markers in differentiation of Mycobacterium bovis strains from animals and humans and for studying epidemiology of bovine tuberculosis. J Clin Microbiol. 1994;32:2425–33.PubMedGoogle Scholar
- Alexander KA, Laver PN, Michel AL, Williams M, van Helden PD, Warren RM, Novel Mycobacterium tuberculosis complex pathogen, M. mungi. Emerg Infect Dis. 2010;16:1296–9. DOIPubMedGoogle Scholar
- Smith NH, Kremer K, Inwald J, Dale J, Driscoll JR, Gordon SV, Ecotypes of the Mycobacterium tuberculosis complex. J Theor Biol. 2006;239:220–5. DOIPubMedGoogle Scholar
- Mostowy S, Inwald J, Gordon S, Martin C, Warren R, Kremer K, Revisiting the evolution of Mycobacterium bovis. J Bacteriol. 2005;187:6386–95. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.PubMedGoogle Scholar
- Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, Proposal for standardization of optimized mycobacterial interspersed repetitive unit–variable-number repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006;44:4498–510. DOIPubMedGoogle Scholar
- Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. DOIPubMedGoogle Scholar
- Rahim Z, Möllers M, te Koppele-Vije A, de Beer J, Zaman K, Matin MA, Characterization of Mycobacterium africanum subtype I among cows in a dairy farm in Bangladesh using spoligotyping. Southeast Asian J Trop Med Public Health. 2007;38:706–13.PubMedGoogle Scholar
- Smith NH, Hewinson RG, Kremer K, Brosch R, Gordon SV. Myths and misconceptions: the origin and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 2009;7:537–44. DOIPubMedGoogle Scholar
- Simeone R, Bottai D, Brosch R. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr Opin Microbiol. 2009;12:4–10. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 18, Number 4—April 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jakko van Ingen, Radboud University Nijmegen Medical Center, Department of Clinical Microbiology (574), PO Box 9101, 6500HB Nijmegen, the Netherlands
Top