Volume 18, Number 4—April 2012
Dispatch
Genomic Analysis of emm59 Group A Streptococcus Invasive Strains, United States
Cite This Article
Citation for Media
Abstract
Genomic analysis of type emm59 group A Streptococcus invasive strains isolated in the United States discovered higher than anticipated genetic heterogeneity among strains and identified a heretofore unrecognized monoclonal cluster of invasive infections in the San Francisco Bay area. Heightened monitoring for a potential shift in the epidemic behavior of emm59 group A Streptococcus is warranted.
Group A Streptococcus (GAS) causes human diseases ranging in severity from uncomplicated pharyngitis to life-threatening necrotizing fasciitis (1). GAS strains have traditionally been classified based on a serologic reaction against the M protein, a polymorphic cell surface adhesin and anti-phagocytic factor; GAS strains are currently typed by sequencing the 5′ hypervariable region of the emm gene, encoding M protein (2–4). Studies of large numbers of GAS isolates causing invasive infections and pharyngitis worldwide have shown that type emm59 GAS strains rarely cause disease (5,6). However, an increase in the frequency and severity of invasive infections caused by type emm59 strains (>500 cases since 2006) has been reported recently in Canada (7). One of the most striking features of the emm59 epidemic in Canada was its rapid spread; invasive emm59 disease was reported in most Canadian provinces and territories in a matter of only a few years (7). By using whole-genome sequencing and animal models of invasive disease, we recently discovered that virtually all type emm59 GAS invasive cases in Canada were caused by a single, recently emerged, hypervirulent emm59 clone (8).
Whole-genome sequence analysis also revealed distinct spatiotemporal patterns of subclone diversification of the epidemic clone in Canada (8). Furthermore, we discovered that several geographically clustered cases of type emm59 GAS invasive infections in south-central Montana in 2010 were caused by a distinct subclone of the emm59 epidemic clone that disseminated from Canada (8). This finding led us to evaluate the hypothesis that the epidemic emm59 clone disseminated further and caused invasive infections in other regions of the United States.
In this study, we sequenced the genomes of all available invasive emm59 GAS strains (m = 40) collected during 2000–2009 by the Active Bacterial Core surveillance (ABCs), a core component of the Centers for Disease Control and Prevention Emerging Infections Programs network. ABCs, an active, laboratory- and population-based surveillance system operating in 10 geographically disparate sites across the United States, represents a population of ≈32 million persons under surveillance for invasive GAS infections (www.cdc.gov/abcs/methodology/surv-pop.html).
Figure A1
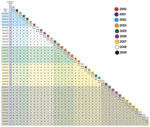
Figure A1. Matrix displaying total number of core genome single-nucleotide polymorphisms separating each individual strain from any other. State and year of isolation are provided for each individual strain. CA, California; CT, Connecticut;...
Genome sequencing was performed by using a Genome Analyzer II (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Polymorphism discovery and phylogenetic analysis were performed as described (8). The reference genome sequence for polymorphism discovery was that of the emm59 Canadian strain MGAS15252 (GenBank accession no. CP003116). On average, the 40 emm59 strains in the ABCs sample differed from the reference strain by 157 single-nucleotide polymorphisms (SNPs) and 15 insertions or deletions. We recently reported that, consistent with our hypothesis, we identified 5 strains that were genetically closely related to the epidemic clone in Canada: 1 strain from Oregon, 2 from California, and 2 from Minnesota (8). Here, we report that the core genomes (i.e., the ≈1,670-kbp portion of the genome that lacks mobile genetic elements and whose gene content is conserved among all sequenced GAS serotypes) of these 5 strains differed from the core genome of reference strain MGAS15252 by <16 SNPs (Figure A1).
Figure
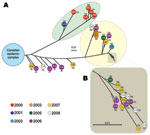
Figure. Inferred genetic relationships among 40 emm59 group A Streptococcus (GAS) strains isolated in the United States during 2000–2009, based on 635 concatenated single-nucleotide polymorphism (SNP) loci identified by whole-genome sequencing. A)...
Although our hypothesis regarding further dissemination of this recently emerged clone into the United States was correct, most emm59 GAS strains collected by the ABCs program were genetically distinct from the clone in Canada. We therefore investigated in more detail the population of emm59 GAS organisms responsible for invasive disease in the United States. We used the whole-genome SNP data to identify 2 major phylogenetic lineages of emm59 organisms (Figure, panel A). All strains recovered during 2000 and 2001 (originating from Minnesota, Maryland, and Georgia) form 1 lineage (Figure, panel A, highlighted in green). On average, the core genomes of these strains differed from that of reference strain MGAS15252 by 141 SNPs. The second lineage consists of 22 strains isolated in California, Connecticut, New Mexico, New York, and Tennessee during 2003–2008 (Figure, panel A, highlighted in yellow). On average, the core genomes of these 22 type emm59 organisms differed from that of reference strain MGAS15252 by 169 SNPs. Seven strains from California, isolated during 2006–2008, were separated from the reference strain by increasing numbers of SNPs (ranging from 36 to 105 SNPs for the most closely to the most distantly related of the 7 strains, respectively) (Figure, panels A).
Closer examination of the second branch of the phylogenetic tree identified a conspicuous group formed by 14 closely related strains isolated from patients in the San Francisco Bay area, California, USA, during 2005–2008 (Figure, panel B). These 14 type emm59 GAS organisms differed from one another, on average, by only 10 SNPs. The phylogenetic and epidemiologic data suggest that these 14 strains constitute a distinct clone that caused a geographic cluster of invasive infections.
The emm59 strains causing the epidemic in Canada were isolated in high percentages from patients with bacteremia and soft tissue infections (7). In the only other well-documented emm59 outbreak, which occurred in Scotland (9), most of the patients had skin lesions and wound infections (9). These data suggest that emm59 GAS strains have a predilection for abscess formation and soft tissue infection. The most common clinical syndromes associated with infections caused by emm59 GAS strains in the ABCs collection were similar to those associated with infections caused by all other emm types (10) and included bacteremia (40%), cellulitis (28%), pneumonia (13%), septic arthritis (5.0%), necrotizing fasciitis (7.5%), and abscess (5.3%). The initial report of type emm59 GAS strains indicated this emm type to be associated with pyoderma and acute glomerulonephritis (11). However, the ABCs program tracks only invasive infections, so it would probably not detect most cases of GAS pyoderma and glomerulonephritis.
In Canada, an epidemiologic association with alcohol abuse, homelessness, hepatitis C virus infection, and illicit drug use was identified, suggesting that the emm59 GAS epidemic was more common among a specific susceptible population consisting primarily of middle-aged persons with underlying medical conditions or histories of substance abuse (7). In our study of type emm59 GAS invasive infection in the United States, we found that 30% of infected persons used illicit drugs and 17.5% abused alcohol; these percentages were higher than those for persons infected with all other emm types (10). However, substance abuse was higher only among US case-patients with infections caused by strains belonging to the epidemic clone from Canada and among case-patients from the San Francisco Bay area. This finding likely reflects circulation of the emm59 strain among subpopulations with similar behaviors. Of note, substance abuse has been shown to be a major risk factor for invasive GAS disease in the San Francisco Bay area (12).
Next-generation DNA sequencing technologies are highly successful for infectious disease epidemiology (13). The emm59 GAS organisms causing invasive infections in the United States were closely related and indistinguishable by multilocus sequence typing. The strains could be differentiated from one another only by the use of high-throughput genome sequencing. The level of genetic diversity we identified among emm59 GAS strains collected by the ABCs program in the United States was considerably greater than that among strains from the epidemic in Canada, where a monoclonal population was found to be responsible for virtually all of the >500 invasive cases reported (8). Of note, the high resolving power of whole-genome sequencing also enabled us to discover a cluster of invasive infections caused by very closely related emm59 GAS strains in the San Francisco Bay area.
Until recently, cases of invasive infection caused by emm59 GAS have been uncommon in North America. However, the recent epidemic in Canada (7,8) and the data presented here for the United States suggest a potential shift in the epidemic behavior of emm59 GAS strains that warrants heightened monitoring awareness by public health authorities.
Dr Fittipaldi is a postdoctoral fellow in the Department of Pathology and Genomic Medicine of the Methodist Hospital Research Institute, Houston, Texas. His primary research interest is the molecular basis of GAS pathogen–host interactions.
Acknowledgments
We acknowledge the Streptococcus Laboratory and emm database (www.cdc.gov/ncidod/biotech/strep/strepblast.htm), Centers for Disease Control and Prevention, for typing and for ABCs isolates. We also thank the following Emerging Infections Programs sites and staff: California Emerging Infections Program; Minnesota Department of Health; and the Emerging Infections Programs in Connecticut, Georgia, Maryland, New Mexico, New York, Oregon, and Tennessee. We are grateful to C.C. Cantu for technical assistance and K. Stockbauer for critical reading of the manuscript.
This work was funded by the Methodist Hospital System. N.F. is funded in part by the Canadian Institutes of Health Research.
References
- Olsen RJ, Shelburne SA, Musser JM. Molecular mechanisms underlying group A streptococcal pathogenesis. Cell Microbiol. 2009;11:1–12. DOIPubMedGoogle Scholar
- Lancefield RC. The antigenic complex of Streptococcus haemolyticus: I. Demonstration of a type-specific substance in extracts of Streptococcus haemolyticus. J Exp Med. 1928;47:91–103. DOIPubMedGoogle Scholar
- Scott JR, Pulliam WM, Hollingshead SK, Fischetti VA. Relationship of M protein genes in group A streptococci. Proc Natl Acad Sci U S A. 1985;82:1822–6. DOIPubMedGoogle Scholar
- Manjula BN, Acharya AS, Fairwell T, Fischetti VA. Antigenic domains of the streptococcal Pep M5 protein. Localization of epitopes crossreactive with type 6 M protein and identification of a hypervariable region of the M molecule. J Exp Med. 1986;163:129–38. DOIPubMedGoogle Scholar
- Luca-Harari B, Darenberg J, Neal S, Siljander T, Strakova L, Tanna A, Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2009;47:1155–65. DOIPubMedGoogle Scholar
- Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9:611–6. DOIPubMedGoogle Scholar
- Tyrrell GJ, Lovgren M, St Jean T, Hoang L, Patrick DM, Horsman G, Epidemic of group A Streptococcus M/emm59 causing invasive disease in Canada. Clin Infect Dis. 2010;51:1290–7. DOIPubMedGoogle Scholar
- Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol. 2012.Epub ahead of print. http://dx.doi.org/10.1016/j.ajpath.2011.12.037DOIPubMedGoogle Scholar
- Phillips G, Efstratiou A, Tanna A, Beall B, Ferguson J, Roworth M. An outbreak of skin sepsis in abattoir workers caused by an ‘unusual’ strain of Streptococcus pyogenes. J Med Microbiol. 2000;49:371–4.PubMedGoogle Scholar
- O’Loughlin RE, Roberson A, Cieslak PR, Lynfield R, Gershman K, Craig A, The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000–2004. Clin Infect Dis. 2007;45:853–62. DOIPubMedGoogle Scholar
- Dillon HC, Dillon MS. New streptococcal serotypes causing pyoderma and acute glomerulonephritis types 59, 60, and 61. Infect Immun. 1974;9:1070–8.PubMedGoogle Scholar
- Passaro DJ, Smith DS, Hett EC, Reingold AL, Daily P, Van Beneden CA, Invasive group A streptococcal infections in the San Francisco Bay area, 1989–99. Epidemiol Infect. 2002;129:471–8. DOIPubMedGoogle Scholar
- Pallen MJ, Loman NJ, Penn CW. High-throughput sequencing and clinical microbiology: progress, opportunities and challenges. Curr Opin Microbiol. 2010;13:625–31. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 18, Number 4—April 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
James M. Musser, Department of Pathology and Genomic Medicine, The Methodist Hospital System, 6565 Fannin St, B490, Houston, TX 77030, USA
Top