Volume 19, Number 3—March 2013
Dispatch
Mycobacterial Lineages Causing Pulmonary and Extrapulmonary Tuberculosis, Ethiopia
Cite This Article
Citation for Media
Abstract
Molecular typing of 964 specimens from patients in Ethiopia with lymph node or pulmonary tuberculosis showed a similar distribution of Mycobacterium tuberculosis strains between the 2 disease manifestations and a minimal role for M. bovis. We report a novel phylogenetic lineage of M. tuberculosis strongly associated with the Horn of Africa.
Ethiopia is among the countries with the highest incidence of tuberculosis (TB) and has a yearly incidence of 261 cases/100,000 population. TB lymphadenitis in cervical lymph nodes (TBLN) accounts for ≈33% of all new cases in this country, which is greater than the global average of ≈15% (1). Ethiopia has the largest livestock population in Africa (≈51 million cattle), and recent studies have shown that bovine TB is endemic in this country (estimated prevalence 1%–10%) (2).
To explore the public health risk for bovine TB in Ethiopia, we have used molecular typing to characterize mycobacterial isolates from persons with TBLN and pulmonary TB who were visiting hospitals throughout the country. Our aim was to define the role of Mycobacterium bovis in human TB and to define the overall structure of the M. tuberculosis complex in Ethiopia.
Patients with suspected TBLN or pulmonary TB who came to hospitals or health centers in study sites and provided voluntary consent were recruited into the study during 2006–2010. Fine needle–aspirate samples and sputum samples were collected from 2,151 patients attending hospitals in Gondar, Woldiya, Ghimbi, Butajira, and Negelle, Ethiopia. In addition, sputum samples were collected from patients at hospitals in Fiche, Jinka, and Filtu and at health centers at 3 suburban sites in Addis Ababa (Holeta, Sululta, and Chancho). Samples were cultured on Löwenstein-Jensen medium supplemented with glycerol or pyruvate and on modified Middlebrook 7H11 medium optimized for culture of M. bovis.
We characterized isolates belonging to the M. tuberculosis complex by using multiplex PCR for large sequence polymorphisms (3,4), spoligotyping (5), and lineage-specific single-nucleotide polymorphism analysis (4,6). Isolates of selected spoligotypes were characterized by 24-loci mycobacterial interspersed repetitive unit–variable number tandem repeat (MIRU-VNTR) analysis (7). Four M. tuberculosis isolates from a group of 36 with unusual spoligotype patterns were further characterized by genome sequencing (Illumina Inc., San Diego, CA, USA). Sequencing reads were mapped to the inferred most recent common ancestor of the M. tuberculosis complex (6). A final alignment of 13,199 single-nucleotide polymorphism positions was generated and analyzed by using the neighbor-joining method with a Tamura-Nei evolutionary model (www.megasoftware.net/mega_papers.php). Nontuberculous mycobacteria were characterized by sequencing of the 16S rDNA gene.
Characteristics of 964 cultures positive for acid-fast bacilli are summarized in Table 1. Most of these isolates had an intact RD9 region, which identified them as M. tuberculosis. Only 4 (0.4%) of 964 isolates had undergone RD9 and RD4 deletions characteristic of M. bovis (3). The 4 M. bovis isolates were obtained from cases of pulmonary TB, 3 of which were from patients living in pastoralist communities in southern Ethiopia. The 10 nontuberculous mycobacterial isolates were identified as M. intracellulare, M. flavescens, and M. simiae; 2 of the isolates were from patients co-infected with M. tuberculosis.
Figure 1
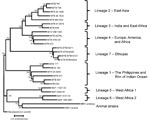
Figure 1. . . Lineages of the Mycobacterium tuberculosis (MTB) complex, Ethiopia, 2006–2010. Genome sequence analysis of 4 strains representative of 36 related isolates identified them as members of a new phylogenetic lineage...
Among the 954 isolates belonging to the M. tuberculosis complex, 671 (71%) belonged to lineage 4, which was the most common lineage in Ethiopia. However, lineage 3 was most prevalent in the northern sites of Gondar and Woldiya (122/257, 47%). Eleven strains belonging to lineage 1 were isolated in the southern region. Two isolates with a characteristic Beijing family spoligotype (spoligotype international type [SIT] 1) were identified as pseudo-Beijing strains belonging to lineage 3 (8). Thirty-six isolates with an unusual spoligotype pattern (missing spacers 4–24) and intact for the TbD1 region could not be assigned to known lineages. Genome sequencing identified these strains as members of a new lineage (lineage 7) localized between ancient lineage 1 and modern lineages 2, 3, and 4 of M. tuberculosis phylogeny (Figure 1). This new lineage 7 was prominent among strains collected in the Woldiya region (17/133 strains, 13%) (Table 1).
Lineage distribution was identical between the 2 disease forms at the national level; lineage 4 was isolated from 71% (442/662) and 70% (229/328) of pulmonary TB and TBLN patients, respectively, and lineage 3 was isolated from 25% (153/622) and 24% (79/328), respectively. The M. tuberculosis isolates encompassed 176 spoligotypes (Technical Appendix Table 1), of which 86 patterns were new to the international genotyping database 2 (SITVIT2) (www.pasteur-guadeloupe.fr:8081/SITVITDemo/) (9). A total of 11% (101/950) of the isolates represented single spoligotypes, and 62% (591/950) were included in 10 major spoligotype clusters (Table 2).
Figure 2
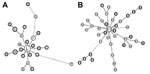
Figure 2. . . Mycobacterial interspersed repetitive unit–variable number tandem repeat (MIRU-VNTR) networks of major spoligotype clusters, Ethiopia, 2006–2010. Two large spoligotype clusters from lineage 4 (A) (90 isolates of sopligotype 149) and...
There was no difference in cluster distribution between pulmonary TB and lymph node TB isolates; 10% and 11%, respectively, were single types, and 64% and 60%, respectively, were included in dominant clusters. Two large clusters representative of lineage 4 (SIT 149) and lineage 3 (SIT 25) were further characterized by MIRU-VNTR typing (Technical Appendix Table 2) and network analysis (Figure 2). In each case, TBLN and pulmonary TB samples were dispersed throughout the network of spoligotype clusters.
All 4 M. bovis isolates from humans showed typical bovine spoligotype profiles lacking spacers 3, 9, 16, and 39–43. In addition, they lacked spacers 4–7 and had deletions of RDAf2, which are features that define strains of the African 2 clonal complex of M. bovis reported from TB-infected cattle in Ethiopia (10).
The frequency of M. bovis in persons in this study (0.4%) is similar to that found in other studies of human TB in Africa (11) and South and Central America (12), but much lower than that observed among selected populations in Tanzania (16%) (13), Ethiopia (17%) (14), and Mexico (28%) (15). These findings indicate that the overall contribution of M. bovis to human TB is minor but greater in specific areas. In Ethiopia, monitoring of zoonotic transmission is needed in urban areas with high rates of bovine TB associated with intensive farming of imported dairy cattle (R. Firdessa et al., unpub. data) and among pastoralist populations from which human M. bovis cases were identified in this study.
Zoonotic transmission of M. bovis can be excluded as the predominant cause of the high national incidence of TBLN in Ethiopia. Mapping of disease networks by spoligotyping and MIRU-VNTR analysis showed an integrated distribution of the 2 disease forms, which suggested that cases of TBLN arise from within the pulmonary TB transmission network, rather than from an external zoonotic source.
We identified a novel phylogenetic lineage of M. tuberculosis (lineage 7) in multiple sites and at a high frequency in Woldiya in the northeastern highlands of Ethiopia. Screening of the SITVIT2 database (9) and the US Centers for Disease Control and Prevention National Tuberculosis Genotyping Surveillance Network Database (L.S. Cowan, pers. comm.) identified 23 (0.03%) of ≈90,000 isolates as members of lineage 7; all were isolated from patients whose country of origin (when known) was in the Horn of Africa. Lineage 7 is of considerable evolutionary interest because it represents a phylogenetic branch intermediate between the ancient and modern lineages of M. tuberculosis (3,4,6).
Dr Firdessa is a doctoral candidate at the University of Würzburg, Würzburg, Germany. His research interest is the molecular epidemiology of tuberculosis.
Acknowledgments
We thank the patients and staff in hospitals and health centers of the participating towns and regions in Ethiopia; the staff at Armauer Hansen Research Institute, and Araya Mengistu for making contributions to this study; and Nalin Rastogi, Thierry Zozio, and David Couvin for interpreting spoligotype patterns and lineage attribution by using the SITVIT2 proprietary database.
This study was supported by the Wellcome Trust, United Kingdom (grant 075833/A/04/Z), under the Animal Health in the Developing World Initiative. The SITVIT2 project was supported by the European Regional Development Fund of the European Commission (ERDF/FEDER, A34-05), and the Regional Council of Guadeloupe (Biodiversity project CR08/031380). The Armauer Hansen Research Institute is supported by the Swedish and Norwegian Development Agencies and the Ethiopian Government. I.C. is supported by the European Union Marie Curie FP7 Actions (project no. 272086), and S.G. is supported by the Swiss National Science Foundation (PP0033_119205).
References
- World Health Organization. Global tuberculosis control: World Health Organization report 2011 [cited 2012 Aug 1]. http://www.who.int/tb/data
- Vordermeier M, Ameni G, Berg S, Bishop R, Robertson BD, Aseffa A, The influence of cattle breed on susceptibility to bovine tuberculosis in Ethiopia. Comp Immunol Microbiol Infect Dis. 2012;35:227–32. DOIPubMedGoogle Scholar
- Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. DOIPubMedGoogle Scholar
- Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008;6:e311. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14 .PubMedGoogle Scholar
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42:498–503. DOIPubMedGoogle Scholar
- Allix-Béguec C, Fauville-Dufaux M, Supply P. Three-year population-based evaluation of standardized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2008;46:1398–406. DOIPubMedGoogle Scholar
- Fenner L, Malla B, Ninet B, Dubuis O, Stucki D, Borrell S, “Pseudo-Beijing”: evidence for convergent evolution in the direct repeat region of Mycobacterium tuberculosis. PLoS ONE. 2011;6:e24737. DOIPubMedGoogle Scholar
- Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. DOIPubMedGoogle Scholar
- Berg S, Garcia-Pelayo MC, Muller B, Hailu E, Asiimwe B, Kremer K, African 2, a clonal complex of Mycobacterium bovis epidemiologically important in East Africa. J Bacteriol. 2011;193:670–8. DOIPubMedGoogle Scholar
- Groenheit R, Ghebremichael S, Svensson J, Rabna P, Colombatti R, Riccardi F, The Guinea-Bissau family of Mycobacterium tuberculosis complex revisited. PLoS ONE. 2011;6:e18601. DOIPubMedGoogle Scholar
- de Kantor IN, Ambroggi M, Poggi S, Morcillo N, Da Silva Telles MA, Osorio Ribeiro M, Human Mycobacterium bovis infection in ten Latin American countries. Tuberculosis (Edinb). 2008;88:358–65. DOIPubMedGoogle Scholar
- Kazwala RR, Daborn CJ, Sharp JM, Kambarage DM, Jiwa SF, Mbembati NA. Isolation of Mycobacterium bovis from human cases of cervical adenitis in Tanzania: a cause for concern? Int J Tuberc Lung Dis. 2001;5:87–91 .PubMedGoogle Scholar
- Kidane D, Olobo JO, Habte A, Negesse Y, Aseffa A, Abate G, Identification of the causative organism of tuberculous lymphadenitis in Ethiopia by PCR. J Clin Microbiol. 2002;40:4230–4. DOIPubMedGoogle Scholar
- Portillo-Gómez L, Sosa-Iglesias EG. Molecular identification of Mycobacterium bovis and the importance of zoonotic tuberculosis in Mexican patients. Int J Tuberc Lung Dis. 2011;15:1409–14. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
2Current affiliation: University of Würzburg, Würzburg, Germany.
Table of Contents – Volume 19, Number 3—March 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Abraham Aseffa, Armauer Hansen Research Institute, All Africa Leprosy Rehabilitation and Training Center Campus, Jimma Rd, PO Box 1005, Addis Ababa, Ethiopia
Top