Volume 19, Number 9—September 2013
Synopsis
Nodding Syndrome
Cite This Article
Citation for Media
Abstract
An epidemic illness characterized by head nodding associated with onchocerciasis has been described in eastern Africa since the early 1960s; we summarize published reports and recent studies. Onset of nodding occurs in previously healthy 5–15-year-old children and is often triggered by eating or cold temperatures and accompanied by cognitive impairment. Its incidence has increased in Uganda and South Sudan over the past 10 years. Four case–control studies identified modest and inconsistent associations. There were nonspecific lesions seen by magnetic resonance imaging, no cerebrospinal fluid inflammation, and markedly abnormal electroencephalography results. Nodding episodes are atonic seizures. Testing has failed to demonstrate associations with trypanosomiasis, cysticercosis, loiasis, lymphatic filariasis, cerebral malaria, measles, prion disease, or novel pathogens; or deficiencies of folate, cobalamin, pyridoxine, retinol, or zinc; or toxicity from mercury, copper, or homocysteine. There is a consistent enigmatic association with onchocerciasis detected by skin snip or serologic analysis. Nodding syndrome is an unexplained epidemic epilepsy.
Nodding syndrome as a distinctive entity was reported from southern Sudan in the 1990s and investigated by local authorities and the World Health Organization (WHO) during 2001–2002 (1,2). In retrospect, children with head nodding, or rhythmic dorsoventral movements of the head (3), as 1 characteristic feature of epilepsy syndromes, had been observed in Tanzania, Liberia, and western Uganda as far back as the 1960s but were not studied separately or described as a distinctive clinical group (3–5). The term nodding disease was first applied in southern Sudan in the 1990s to describe the occurrence of repetitive head nodding, characteristically occurring among children while eating, and variably associated with other seizure activity, neurologic and cognitive impairment, delayed puberty, and growth retardation.
Thousands of cases have been reported from southern Tanzania, northern Uganda, and South Sudan, although much smaller numbers have been documented and investigated in any detail (6–13). The effect of the disease on families and communities can be devastating because previously healthy young children drop out of school, lose the ability to eat, and require constant oversight because they might fall into a cooking fire or wander off and drown. Local authorities and national governments requested assistance from WHO, the US Centers for Disease Control and Prevention (CDC), and other agencies.
Figure 1
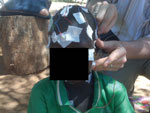
Figure 1. . . Child with nodding syndrome, on whom electroencephalographic leads are being attached, Uganda, 2009.
Investigations have confirmed a similar syndrome in Uganda and southern Sudan, in which the syndrome produced the characteristic clinical features, the age of onset was tightly clustered among children 5–15 years of age, and the reported incidence became higher during recent years (Figure 1) (7,10–14). Case series of patients have been intensively described and investigated by evaluations of cerebrospinal fluid (CSF), brain imaging, and video electroencephalography (EEG), and 4 case–control studies have been conducted to assess risk factors for the disease and test for infectious pathogens, toxin exposures, and nutritional deficiencies (2,6–10). Associations with onchocerciasis and nutritional deficiencies have been consistent features, but no definitive underlying cause has been identified.
Major unanswered questions remain about the reason for the persistent association with onchocerciasis, possible contributions of nutritional deficiencies or unidentified toxin exposures, and optimal treatment and prognosis. Some of the most detailed investigations have been conducted recently or are ongoing, and much of what is known about the syndrome remains unpublished. In this review, we aimed to include all available information, including unpublished reports of earlier investigations and major recent findings, to provide the fullest possible picture (Table 1).
According to media reports and assessments from local officials, there may be as many as 3,000–8,000 cases of nodding syndrome in the districts of Kitgum, Pader, and Lamwo in northern Uganda, and Western and Central Equatoria States in South Sudan (1,15). In March 2012, the Uganda government actively sought to register cases for the purpose of providing services and recorded >3,000 names, although a standardized and consistently applied case definition was not used. Detailed investigations that used consistently applied case definitions and active community outreach have identified ≥224 cases across Kitgum District in Uganda and 260 cases in Western Equatoria State in South Sudan; complete ascertainment of all cases was not the primary objective in either investigation (6,8,11). Widespread registration of cases has not been completed in Tanzania, but in a study conducted in 2005, a total of 62 cases were documented and investigated in detail (9). What seems clear is that there are at least several hundred affected children currently in the 3 geographic areas, and the actual numbers might be much higher. In July 2012 a standard set of case definitions (Table 2) was developed during an international meeting on nodding syndrome in Kampala, Uganda (16), and applied in an extensive community survey across affected districts of northern Uganda.
Deaths among nodding syndrome patients also are commonly reported but incompletely ascertained. A Uganda news report in May 2012 listed the number of deaths at 205, but the Ministry of Health could not confirm that all of these deaths were a result of nodding disease (15). Anecdotal reports of deaths from drowning, burns, and other causes among nodding syndrome patients are common, but a collaborative effort between the Uganda Ministry of Health and CDC to register deaths and obtain autopsy specimens resulted in 1 autopsy over an 18-month period during 2011–2012. A 2009 follow-up investigation of 62 patients with nodding syndrome in Tanzania first evaluated in 2005 identified 2 deaths that occurred in the interim (9,10). A follow-up investigation of 12 patients in Uganda evaluated in 2009 and 2010 identified interval worsening in 6 patients, improvement in none, and no deaths (7). Although the mortality rate associated with nodding syndrome remains to be accurately defined, media reports from affected communities imply that this rate is high, and long-term studies of childhood epilepsy also suggest that it will probably increase (17).
Figure 2
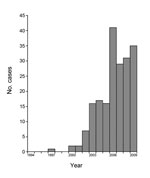
On the basis of reports for Uganda and probably for South Sudan, the incidence of nodding syndrome appears to be increasing (Figure 2). The earliest cases among the 224 documented patients in Uganda occurred in 2000, except for 1 possible onset in 1997, and there was a steady increase in cases identified through the study in 2009. In South Sudan, the earliest cases recognized were in 1991 in Mundri County and 1995 in Lui Township. Community reports from a village administrator in Sudan (1) and focus groups in Uganda (6) indicated that previous generations had not been affected by this disease. In contrast, reports of head nodding in Tanzania date back >50 years (4), and it is not clear from available reports whether the incidence has increased.
Figure 3
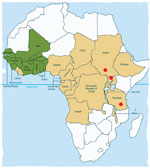
Figure 3. . . Countries in the former Onchocerciasis Control Programme in western Africa in which onchocerciasis was eliminated as a public health problem through vector control (green); countries in the African Programme...
The disease appears to be localized in 3 noncontiguous areas in South Sudan, Uganda, and Tanzania (Figure 3). Although head nodding as 1 feature of seizure disorders has been reported from Liberia (3), Taiwan (18), and elsewhere, clustering of hundreds of cases of this syndrome and the same manifestations has not been described elsewhere. Although onchocerciasis is endemic to all 3 areas, the distribution of this parasitic disease is much wider, extending to across much of eastern and western Africa (19,20) and Central and South America (21), which are huge areas with populations apparently unaffected by nodding syndrome (Figure 3).
Most of the populations affected by nodding syndrome were internally displaced; in Uganda and Sudan, the conflict with the Lord’s Resistance Army during the 1990s resulted in dependence on refugee camps and in widespread food shortages during the years preceding nodding syndrome. In Tanzania, most (58/62) of the described patients with nodding syndrome were members of the Pogoro tribe (9). Although the Pogoro were not recently internally displaced refugees, they are among the poorest of the region, and therefore susceptible to food shortages (22). Potential associations of nodding syndrome with hunger, specific micronutrient malnutrition, or ingestion of toxic substances or contaminated relief foods have been explored in 4 case–control studies, as detailed below.
Figure 4
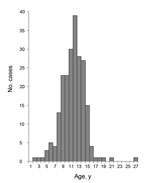
Figure 4. . . Age distribution of patients at onset of nodding syndrome, Kitgum District, Uganda. For nodding syndrome elsewhere, age distribution tightly clusters in persons 5–15 years of age. Modified from Foltz...
The distinctive age distribution (tight clustering among persons 5–15 years of age) is a consistent feature of nodding syndrome (Figure 4). Caregivers report that the children were unaffected as infants and had apparently normal growth; most of these children achieved their developmental milestones until the onset of nodding (6,8,10). Although persons of other ages with onset of nodding are occasionally identified, the disease is rare among younger children or adults. Onset in late childhood or early adolescence is seen in certain epilepsy syndromes (3,5), autoimmune diseases such as juvenile idiopathic arthritis (23), Sydenham chorea and other complications of group A streptococcal infections (24), and some nutritional toxicities such as konzo (25) and neurolathyrism (26). Many epidemic infectious diseases predispose the very young or elderly, but some clustering of infections among persons 5–15 years of age are occasionally seen in neurologic infections such as meningococcal meningitis (27), parasitic infections such as urinary schistosomiasis (28), or epidemic viral infections such as mumps (29).
Clinical evaluations of nodding disease as a distinctive entity began with the more formal description of the syndrome in South Sudan in the late 1990s and have included at least 4 detailed investigations (Table 3). Because case definitions were not standardized, studies might include clinical entities that were not identical, but repetitive head nodding was common to all studies, and definitions used were broadly compatible with the new standards (Table 2).
General clinical descriptions identify variations in behavior and body habitus, ranging from children who appeared and behaved appropriately for age to those that were obtunded, drooling, and unable to stand or walk independently. Stunting and wasting were commonly documented, as were facial scars from burns and other injuries. It is not known whether stunting and wasting are results of the disease or predisposing factors. Dysmorphic facial features, chest wall abnormalities, dwarfism, and delayed sexual development have been noted but not consistently documented.
Focal motor abnormalities were found in a few of the 81 patients who were given complete examinations, including upper motor neuron abnormalities in 4 patients (9), ataxia in 1 (7), and involuntary movements and nystagmus in 9 (2). Clinical findings often associated with epileptic encephalopathy, such as altered level of consciousness, drooling, and incontinence of urine, were noted more frequently (2,7).
In contrast, all 4 investigations identified major cognitive deficits, despite the challenges of standardized testing in the rural setting in Africa. In early investigations in Sudan, 15% of patients had mental retardation (test not specified) (2). Of the 62 persons in Tanzania, 40% had cognitive impairment (as reported by mothers who used a simple grading system of slight, moderate, and severe impairment), half of them severe. In Uganda, a simple neurocognitive instrument administered to 78 case-patients and age-matched controls showed major cognitive impairment among case-patients (7). Anecdotally, cognitive impairment appears to be progressive (2,7). Evidence of psychiatric disturbances have included visual and auditory hallucinations reported in 29% and 26% of patients, respectively (6), along with occasional features such as shouting, screaming, and jumping up and running in circles (2).
Nodding episodes occur several times a week to many times per day, and episodes have been observed by investigators, recorded videographically, and documented by video–EEG (7). Often triggered by eating or cold weather, the head drops repeatedly toward the chest in cycles of 5–20 nods/min for several minutes. Nodding may be accompanied by automatisms or other seizure-like activity, and the child is either unaware of his or her surroundings or has a decreased ability to continue an activity (e.g., eating) or respond to questions or external stimuli.
Figure 5

Figure 5. . . Ictal electroencephalographic recording of a 12-year-old boy in Uganda with nodding syndrome obtained during a typical nodding episode. Shown is a sudden electrodecremental episode with concomitant electromyographic decrease in...
Simultaneous recording of 2 episodes by EEG, videography, electromyography, and electrocardiography documented that the nodding episodes are manifestations of atonic seizures. In these case-patients, head nodding was associated with generalized electrodecrement, followed by generalized sharply contoured rhythmic theta activity, dropping of the chin, and paraspinal electromyographic abnormalities (Figure 5) (7). Other EEG recordings in ≥61 patients reported or observed to have nodding syndrome have consistently identified interictal abnormalities and various electrographic seizure types (2,7,9). In the Sudan patient series, which included 32 patients at various stages of disease, worsening or more severe clinical disease was associated with more severe EEG findings, as shown by progressively more abnormal background activity, ultimately resulting in diffuse subcontinuous nonreacting delta–theta activity and loss of normal cerebral electrical architecture (2). Biphasic or triphasic periodic sharp waves of the type frequently seen in human prion diseases or metabolic disorders were not observed in any of these assessments, and there were no periodic lateralizing epileptiform discharges suggestive of encephalitis from herpesvirus or other viruses.
Assessments of CSF have been documented for ≥ 80 patients in Tanzania, South Sudan, and Uganda (2,7,9). Specimens were generally characterized as grossly clear, and all glucose and protein levels were within reference ranges for age. Among 48 patients for whom CSF cell counts were available, only 3 (6%) patients had increased leukocyte counts of 6, 8, and 28 cells/μL, and the third sample was reported as being contaminated with blood (9).
Figure 6
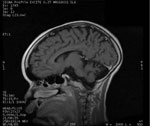
Figure 6. . . Magnetic resonance image of a 13-year-old boy in Uganda with nodding syndrome. Image shows prominent cortical atrophy.
Brain magnetic resonance imaging has been documented for ≥17 patients in Tanzania and Uganda (7,9). For 4 of the patients in Tanzania, images were interpreted as showing normal results. Images for all 5 patients in Uganda were interpreted as showing generalized atrophy (Figure 6). Five patients from Tanzania had nonspecific signal abnormalities with hyperintensity on T2-weighted images, and 5 patients from Tanzania and 2 from Uganda had hippocampus abnormalities. None of the patients showed evidence of meningeal or parenchymal inflammation, cystic or other lesions consistent with acute disseminated encephalomyelitis, neurocysticercosis, central nervous system tuberculosis, or other focal brain infection. Pulvinar sign (30), cortical ribboning (31), or other findings suggestive of human prion disease have not been identified.
One brain from a patient with nodding syndrome in Uganda was examined pathologically at Makerere University (Kampala, Uganda) and at CDC (Atlanta, GA, USA). Because of delays in obtaining the autopsy and fixation of the brain, tissue sections were degraded and largely uninterpretable.
The response to different anti-epileptic drugs has been variably reported by parents and clinicians as occasionally but not consistently helpful. Winkler et al. characterized seizure control among 59 patients receiving therapy (primarily phenytoin and phenobarbital) as being effective compared with seizure control before treatment, but 45% still had head nodding 1–5 times/month, and 21% still had daily episodes (10). Similar impressions were reported from caretakers and parents in Uganda (7) and Sudan (2). The comparative efficacy of anti-epileptic drugs has not been assessed in controlled studies.
Two general approaches for identification of the underlying cause of nodding syndrome have been taken. The first approach is laboratory testing of a series of patients for various infectious agents, toxin exposures, or nutritional deficiencies (2,6,8,9). The second approach is comparison of laboratory-based or reported exposures of such patients with a group of unaffected control children of similar ages from the same location (2,6,8).
Four case–control studies have been performed among 173 case-patients and 179 controls (Table 4). Dozens of exposures have been assessed by history, physical examination, or laboratory testing; only significantly associated exposures or prominent a priori hypotheses are shown in Table 4. Several exposures have been moderately more common among case-patients, and none of the findings except for results of tests for infection with Onchocerca volvulus nematodes has been reproducible across various investigations (Tables 5, 6).
In the early investigations from Sudan, consumption of red sorghum, consumption of baboon brain, and absence of reported history of measles were identified as being potentially associated with nodding syndrome, but assessments in the 2 studies in Uganda and the subsequent study in Sudan did not confirm these findings. Consumption of crushed roots in Uganda in 2009 was also not confirmed as a pre-illness exposure when explored with additional questions in 2010 and 2011. The association with munitions in Uganda in 2009 was found to be an association with gun raids and not chemicals, as initially hypothesized. In addition to associations with microfilariae obtained by skin snips and serum antibodies against 3 recombinant Onchocerca spp. nematode–specific proteins (Ov-16, Ov-FAR1, and Ov-MSA), the observation that case-patients were more likely than controls to show stunting and wasting was a consistent finding, as was a prominent deficiency of pyridoxine (vitamin B6) among case-patients and controls in Uganda and South Sudan.
Testing in these studies and observational series of case-patients in Tanzania has been extensive. Results can be grouped into those for tests for known or hypothesized causes of infectious encephalitis or postinfectious encephalopathy (Table 5), and toxic encephalopathy, nutritional neuropathologic changes, or genetic epilepsy disorders (Table 6). A molecular approach that does not require a priori hypotheses and uses broadly reactive PCR primers specific for 19 viral families and represents hundreds of potential pathogens, has shown negative results (6). Most of the hypothesis-directed testing has also shown negative results.
The various tests for onchocerciasis are notable exceptions to these findings. In all studies that assessed the association between onchocerciasis and nodding syndrome, there has been a trend toward more positive results for case-patients than controls, and this trend has been observed in testing for microfilaria by skin snip in the 2 studies in South Sudan (2,8) and in testing for serum antibodies against recombinant Onchocerca spp.–specific proteins in the 2 studies in Uganda (6). Skin snip positivity is strongly reduced by treatment with ivermectin (32), but 2 comparisons of case-patients and controls given ivermectin failed to show that case-patients were less likely to have been treated (Table 4). The assumption has been that microfilariae seen were those of O. volvulus nematodes and that antibodies are against Onchocerca spp. microfilariae, but these 2 assumptions might be inconsistent with some reported results. The first result is that PCRs for spinal fluid from 48 patients in Tanzania and 16 in South Sudan were negative for O. volvulus nematodes (2,9). The second result is that sequencing of DNA from a limited number of skin snip specimens from Uganda suggests the presence of an organism closely related to Mansonella spp. nematodes.
The underlying cause of nodding syndrome is a mystery. Studies summarized in this report, taken collectively, have evaluated and eliminated dozens of reasonable hypotheses, including unknown pathogens. Documentation of pathogenesis should be helpful in narrowing the list of possibilities, but identifying the cause of this or any novel epilepsy syndrome is likely to remain a challenge.
The persistent association of nodding syndrome with onchocerciasis is puzzling. Onchocerciasis is widely distributed in areas that do not have nodding syndrome or, considering that systematic evaluations have not been undertaken, where nodding syndrome is not prevalent enough to have resulted in awareness/reporting of the syndrome. This finding suggests an unidentified cofactor or a variant strain of the organism. The possible role of onchocerciasis in epilepsy is an issue of ongoing debate (33–36), but the organism is not believed to be neuroinvasive, and negative PCR results for 64 CSF specimens from Tanzania and South Sudan further substantiates this suggestion. Recent findings of DNA sequencing of skin snip specimens from Uganda raised additional questions and may point toward a morphologically similar and antigenically cross-reactive filarial species.
Pyridoxine deficiency has been a consistent and unexpected finding among case-patients and controls tested, which is notable because of the known association between abnormal pyridoxine metabolism and complex, intractable seizures (37,38). Pyridoxine-dependent seizure is a genetic disease that is not clinically consistent with nodding syndrome, but infants with pyridoxine-dependent seizures can be treated and cured with high daily doses of oral pyridoxine (38). A clinical trial is planned in Uganda to assess the role of high-dose pyridoxine, as well as conventional anti-epileptic medications, as treatment for nodding syndrome. Careful evaluation of the relationship between onchocerciasis and pyridoxine in the investigations in Uganda and South Sudan has failed to document an interaction between the 2 variables.
Nutritional toxicity remains a possible cause of nodding syndromes. Konzo, a neurologic disorder that causes permanent spastic paraparesis, shares several epidemiologic similarities to nodding syndrome, including narrow age group clustering among persons 5–15 years of age (25). Konzo results from consumption of improperly prepared species of cassava and is sometimes seen during times of famine and particularly among persons with dietary deficiencies of sulfur-containing amino acids. For nodding syndrome, a lack of association of cassava consumption in case–control studies and negative results of testing for urinary thiocyanate indicate that acute or ongoing cyanate exposure makes this specific etiology less likely. However, a similar but unrecognized form of nutritional toxicity remains a possible cause.
Neuronal antibodies represent 1 possible common pathway for a novel epidemic epilepsy associated with exposure to O. volvulus nematodes. Such a mechanism is recognized in Sydenham chorea, a distinctive movement disorder now known to result from neuronal antibodies produced against group A streptococcal antigens but cross-reacting with neuronal epitopes in basal ganglia (39). Why only a tiny fraction of those with a streptococcal throat infection end up with chorea remains a mystery. In recent years, the number of characterized neuronal antibodies has increased for those antibodies affecting a limited number of patients with paraneoplastic syndromes to a broader range of known antibodies and clinical manifestations (40). A recent description of 32 patients with autoimmune epilepsy highlighted several distinguishing features reminiscent of features of nodding syndrome, including frequent complex partial seizures refractory to anti-epileptic drugs, prominent cognitive compromise, and occasional psychiatric symptoms (40). Although preliminary testing for known neuronal antibodies at the Mayo Clinic (Rochester, MN, USA) (V. Lennon, unpub. data) and preliminary screening for known or unknown antibodies at Emory University (Atlanta, GA, USA) (A. Levey, unpub. data) have not identified such antibodies in specimens from patients with nodding syndrome, this possibility remains under active investigation.
Aside from questions regarding the underlying cause of nodding syndrome, many questions remain regarding the long-term course, prognosis, optimal treatment, and effective approaches for families and communities. The long-term mortality rate and how to improve survival times and rates remain purely speculative. Reports from parents and clinicians regarding effectiveness of different anti-epileptic drugs are anecdotal with rare exceptions (10), and a controlled trial of treatment would be invaluable. Affected families and communities will probably be coping with chronically dependent patients with nodding syndrome for many years with few resources and considerable pressure for effective management.
Acknowledgments
We thank the families and communities for ongoing support of investigations; Allan Levey and Vanda Lennon for sharing unpublished observations; Joseph Kubofcik, Martha Anker, Stella Chungong, Mickey Richer, Peter Spencer, James Tumwine, Martin Opoka, Ayana Yeneabat, Andrea Quarello, Doug Klaucke, Jeffrey Ratto, Carlos Navarro Colorado, Christina Gurnett, Lorraine Alexander, Louise Jilek-Aall, and other members of the investigative teams from South Sudan, Uganda, and Tanzania for assistance; and WHO for international leadership and coordination.
Dr Dowell is director of the Division of Global Disease Detection and Emergency Response at CDC, Atlanta, GA. His research interests focus on the causes of enigmatic disease outbreaks.
References
- Lacey M. Nodding disease: mystery of southern Sudan. Lancet Neurol. 2003;2:714 and. DOIPubMedGoogle Scholar
- Tumwine JK, Vandemaele K, Chungong S, Richer M, Anker M, Ayana Y, Clinical and epidemiologic characteristics of nodding syndrome in Mundri County, South Sudan. Afr Health Sci. 2012;12:242–8 .PubMedGoogle Scholar
- van der Waals F, Goudsmit J, Gajdusek DC. See-ee: clinical characteristics of highly prevalent seizure disorders in the Gbawein and Wroughbarh clan region of Grand Bassa County, Liberia. Neuroepidemiology. 1983;2:35–44. DOIGoogle Scholar
- Aall-Jilek LM. Epilepsy in the Wapogoro tribe in Tanganyika. Acta Psychiatr Scand. 1965;41:57–86. DOIGoogle Scholar
- Kaiser C, Benninger C, Asaba G, Mugisa C, Kabagambe G, Kipp W, Clinical and electro-clinical classification of epileptic seizure in west Uganda. Bull Soc Pathol Exot. 2000;93:255–9 .PubMedGoogle Scholar
- Foltz J, Makumbi I, Sejvar JJ, Malimbo M, Ndyomugyenyi R, Atai-Omoruto AD, An epidemiologic investigation of potential risk factors for nodding syndrome in Kitgum District, Uganda. PLoS ONE. 2013;8. In press. DOIPubMedGoogle Scholar
- Sejvar JJ, Kakooza AM, Foltz JL, Makumbi I, Atai-Omoruto AD, Malimbo M, Clinical, neurologic, and electrophysiologic features of nodding syndrome, Kitgum, Uganda: an observational case series. Lancet Neurol. 2013;12:166–74 . DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Nodding syndrome—South Sudan, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:52–4 .PubMedGoogle Scholar
- Winkler AS, Friedrich K, Konig R, Meindl M, Helbrok R, Unterberger I, The head nodding syndrome: clinical classification and possible causes. Epilepsia. 2008;49:2008–15 . DOIPubMedGoogle Scholar
- Winkler AS, Friedrich K, Meindl M, Kidunda A, Nassri A, Jilek-Aall L, Clinical characteristics of people with head nodding in southern Tanzania. Trop Doct. 2010;40:173–5. DOIPubMedGoogle Scholar
- Nyungura JL, Lako A, Gordon A, Lejeng L, William G. Investigation into the nodding syndrome in Witto Payam, Western Equatoria State, 2010. Southern Sudan Medical Journal. 2011;4:3–6.
- Donnelly J. CDC planning trial for mysterious nodding syndrome. Lancet. 2012;379:299. DOIPubMedGoogle Scholar
- Williams SC. Nodding syndrome leaves baffled scientists shaking their heads. Nat Med. 2012;18:334 . DOIPubMedGoogle Scholar
- Wasswa H. Ugandan authorities deal with a mysterious ailment that leaves people nodding continuously. BMJ. 2012;344:e349. DOIPubMedGoogle Scholar
- Fallon A. Hundreds of new nodding disease cases reported. iUganda 2012;Section. Uganda local news story [cited 2013 Jun 4]. http://iuganda.ug/news/local/11793-hundreds-of-new-nodding-cases-reported.html
- World Health Organization. Report of an international conference on nodding syndrome. Bull World Health Organ. 2013;91. In press.
- Sillanpää M, Shinnar S. Long-term mortality in childhood-onset epilepsy. N Engl J Med. 2010;363:2522–9. DOIPubMedGoogle Scholar
- Lin FC, Wei LJ, Shih PY. Effect of levetiracetam on truncal tic in neuroacanthocytosis. Acta Neurol Taiwan. 2006;15:38–42 .PubMedGoogle Scholar
- Noma M, Nwoke BE, Nutall I, Tambala PA, Enyong P, Namsenmo A, Rapid epidemiological mapping of onchocerciasis (REMO): its application by the African Programme for Onchocerciasis Control (APOC). Ann Trop Med Parasitol. 2002;96(Suppl 1):S29–39. DOIPubMedGoogle Scholar
- World Health Organization Economic and Social Council. Onchocerciasis and its control. Geneva: The Organization; 1995.
- Sauerbrey M. The Onchocerciasis Elimination Program for the Americas (OEPA). Ann Trop Med Parasitol. 2008;102(Suppl 1):25–9 . DOIPubMedGoogle Scholar
- Jilek-Aall L, Jilek W, Miller JR. Clinical and genetic aspects of seizure disorders prevalent in an isolated African population. Epilepsia. 1979;20:613–22. DOIPubMedGoogle Scholar
- Demirkaya E, Ozen S, Bilginer Y, Ayaz NA, Makay BB, Unsal E, The distribution of juvenile idiopathic arthritis in the eastern Mediterranean: results from the registry of the Turkish Paediatric Rheumatology Association. Clin Exp Rheumatol. 2011;29:111–6 .PubMedGoogle Scholar
- Demiroren K, Yavuz H, Cam L, Oran B, Karaaslan S, Demiroren S. Sydenham’s chorea: a clinical follow-up of 65 patients. J Child Neurol. 2007;22:550–4. DOIPubMedGoogle Scholar
- Bonmarin I, Nunga M, Perea WA. Konzo outbreak, in the south-west of the Democratic Republic of Congo, 1996. J Trop Pediatr. 2002;48:234–8. DOIPubMedGoogle Scholar
- Getahun H, Mekonnen A. TekleHaimanot R, Lambein F. Epidemic of neurolathyrism in Ethiopia. Lancet. 1999;354:306–7. DOIPubMedGoogle Scholar
- Mutonga DM, Pimentel G, Muindi J, Nzioka C, Mutiso J, Klena JD, Epidemiology and risk factors for serogroup X meningococcal meningitis during an outbreak in western Kenya, 2005–2006. Am J Trop Med Hyg. 2009;80:619–24 .PubMedGoogle Scholar
- Morenikej OA, Idowu BA. Studies on the prevalence of urinary schistosomiasis in Ogun State, south-western Nigeria. West Afr J Med. 2011;30:62–5 .PubMedGoogle Scholar
- Barskey AE, Glasser JW, LeBaron CW. Mumps resurgences in the United States: a historical perspective on unexpected elements. Vaccine. 2009;27:6186–95. DOIPubMedGoogle Scholar
- Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M, Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol. 2003;24:1560–9 .PubMedGoogle Scholar
- Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol. 2008;64:97–108 . DOIPubMedGoogle Scholar
- Basáñez MG, Pion SD, Boakes E, Filipe JA, Churcher TS, Boussinesq M. Effect of single-dose ivermectin on Onchocerca volvulus: a systematic review and meta-analysis. Lancet Infect Dis. 2008;8:310–22 . DOIPubMedGoogle Scholar
- Pion SD, Kaiser C, Boutros-Toni F, Cournil A, Taylor MM, Meredith SE, Epilepsy in onchocerciasis endemic areas: systematic review and meta-analysis of population-based surveys. PLoS Negl Trop Dis. 2009;3:e461 . DOIPubMedGoogle Scholar
- Kaiser C, Rubaale T, Tukesiga E, Kipp W, Kabasambe G, Ojony JO, Association between onchocerciasis and epilepsy in the Itwara hyperendemic focus, West Uganda: controlling for time and intensity of exposure. Am J Trop Med Hyg. 2011;85:225–8. DOIPubMedGoogle Scholar
- Katabarwa M, Lakwo T, Habumogisha P, Richards F, Eberhard M. Could neurocysticercosis be the cause of “onchocerciasis-associated” epileptic seizures? Am J Trop Med Hyg. 2008;78:400–1 .PubMedGoogle Scholar
- König R, Nassri A, Meindl M, Matuja W, Kidunda AR, Siegmund V, The role of Onchocerca volvulus in the development of epilepsy in a rural area of Tanzania. Parasitology. 2010;137:1559–68. DOIPubMedGoogle Scholar
- Stockler S, Plecko B, Gospe SM Jr, Coulter-Mackie M, Connolly M, van Karnebeek C, Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol Genet Metab. 2011;104:48–60. DOIPubMedGoogle Scholar
- Gospe SM Jr. Neonatal vitamin-responsive epileptic encephalopathies. Chang Gung Med J. 2010;33:1–12 .PubMedGoogle Scholar
- Kirvan CA, Swedo SE, Kurahara D, Cunningham MW. Streptococcal mimicry and antibody-mediated cell signaling in the pathogenesis of Sydenham's chorea. Autoimmunity. 2006;39:21–9. DOIPubMedGoogle Scholar
- Quek AM, Britton JW, McKeon A, So SE, Lennon VA, Shin C, Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69:582–93. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 19, Number 9—September 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Scott F. Dowell, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop D69, Atlanta, GA 30333, USA
Top