Volume 20, Number 11—November 2014
Research
Novel Chlamydia trachomatis Strains in Heterosexual Sex Partners, Indianapolis, Indiana, USA
Cite This Article
Citation for Media
Abstract
Chlamydia trachomatis causes a high number of sexually transmitted infections worldwide, but reproducible and precise strain typing to link partners is lacking. We evaluated multilocus sequence typing (MLST) for this purpose by detecting sequence types (STs) concordant for the ompA genotype, a single-locus typing standard. We tested samples collected during April 2000–October 2003 from members of established heterosexual partnerships (dyads) in the Indianapolis, Indiana, USA, area who self-reported being coital partners within the previous 30 days. C. trachomatis DNA from 28 dyads was tested by MLST; sequences were aligned and analyzed for ST and phylogenetic relationships. MLST detected 9 C. trachomatis STs, 4 unique to Indianapolis; STs were identical within each dyad. Thirteen unique strains were identified; 9 (32%) dyads harbored novel recombinant strains that phylogenetically clustered with strains comprising the recombinants. The high rate of novel C. trachomatis recombinants identified supports the use of MLST for transmission and strain diversity studies among at-risk populations.
Chlamydia trachomatis, a bacterium that can infect both men and women, is most commonly sexually transmitted. In 2008, approximately 105.7 million new C. trachomatis sexually transmitted infections (STIs) occurred worldwide (1); an estimated 2.86 million incident cases occurred in the United States (2). The last surveillance study of STIs in the United States, in 2011, reported 1,412,791 chlamydial infections, the largest case number for any disease ever reported to the Centers for Disease Control and Prevention (3).
C. trachomatis infections in men and women are mostly asymptomatic; thus, continued sexual activity among persons unaware of their infection status facilitates further transmission. Gaps in knowledge of chlamydial STIs include how measures of immunity, bacterial load, condom use, and other factors relate to transmission risk. Longitudinal studies of these factors are needed to inform treatment and prevention strategies (4). The tools required include careful ascertainment of sexual history and behavioral determinants and reproducible and discriminating biomarkers to strengthen the case for transmission between sex partners linked by partner tracing.
The standard biomarker for these studies is ompA genotyping, but this method lacks precision because the gene is under immune selection and represents only 0.1% of the genome. Because large-scale whole-genome sequencing of clinical samples is not yet feasible, multilocus sequence typing (MLST) for C. trachomatis has been developed to provide greater insight into strain types; 3 such MLST methods have been reported in the literature (5–10). The scheme we developed, on the basis of analysis of 19 reference strains and 68 geographically diverse clinical isolates, identified 44 MLST sequence types (STs), compared with only 20 ompA genotypes (11). In our scheme, we were also able to discriminate single-nucleotide polymorphisms (SNPs) that correlate with disease phenotypes attributable to C. trachomatis: lymphogranuloma venereum (LGV), trachoma, and non-LGV urogenital diseases (11). Our scheme has since been expanded to encompass 192 geographically and clinically diverse samples.
For this study, we applied our MLST scheme to a subset of a well-defined heterosexual partnership (dyad) cohort in Indianapolis, Indiana, USA, comprising 28 dyads for which concordance of the ompA genotype existed between partners. The purpose of the study was to determine whether MLST, which provides a more detailed level of strain typing than ompA genotyping, would also show strain concordance between partners, as would be expected if transmission had occurred within the dyads. In addition, we sought to identify additional C. trachomatis strain types, beyond those identified by ompA genotyping, that might be unique to Indianapolis, because this geographic region has not previously been included in any MLST database.
Study Population
A study of C. trachomatis concordance in heterosexual partnerships (dyads) was conducted in Indianapolis during April 2000–October 2003; participants were sexually active heterosexual men and women 15–25 years of age who visited an urban STI clinic (12). Written informed consent was obtained, and the study was approved by the Indiana University–Purdue University Institutional Review Board. Eligibility was defined as self-reported sexual activity between the partners during the previous 30 days. A total of 210 heterosexual dyads were established by research disease intervention specialists and enrolled. C. trachomatis infection was identified by Amplicor CT/NG (Roche Diagnostics, Indianapolis, IN, USA) nucleic acid amplification test and cell culture, as previously described (12). Of the 210 dyads, 130 contained >1 C. trachomatis–infected partner; for 45 dyads, both partners were infected and had identical ompA genotypes.
For the MLST study, we used remainder samples from 56 members of 28 dyads who were concordant for C. trachomatis infection and ompA genotype. These samples were provided to investigators at Children’s Hospital Oakland Research Institute (CHORI) in a de-identified and blinded fashion. Thus, CHORI research was considered not to involve human subjects, and informed consent was not required.
Reference and Clinical Samples
We used 56 samples (from cervix in women and urethra in men) from 28 dyads in which persons within each dyad were concordant for C. trachomatis infection and ompA genotype. Additionally, we used for analysis MLST data for 20 C. trachomatis reference strains (A/Sa1, A/HAR13, B/TW5/OT, Ba/Apache2, C/TW3/OT, D/UW3/Cx, Da/TW448, E/Bour, F/ICCal3, G/UW57/Cx, H/UW4/Cx, I/UW12/Ur, Ia/UW202, J/UW36/Cx, Ja/UW92, K/UW36/Cx, L1/440, L2/434, L2a/UW396, L3/404) and 172 clinical samples in the MLST database (http://www.mlst.net).
ompA Genotyping and MLST Analyses
ompA genotyping of the samples had been previously performed at Indiana University (13,14) as part of the earlier C. trachomatis concordance study. Cultured and noncultured clinical samples were sent to CHORI for analysis (11). DNA was extracted, and MLST for 7 housekeeping genes was performed by using primers as described (11; http://www.mlst.net; Technical Appendix Table 1). A consensus sequence was created from forward and reverse sequences, and the genes were concatenated and queried against all 202 MLST sequences in the database (15). Sequence output was used to identify each unique allelic profile to assign an ST, and all STs were deposited in the C. trachomatis database (http://chlamydia.mlst.net). The concatenated sequences of the 7 MLST loci and the allelic profiles for each sample were used to identify sample relatedness.
ompA genotypes were defined on the basis of homology with reference strains of C. trachomatis. If >1 SNP was identified when sequences were compared to those of the closest hit reference strain, a number was used to denote the presence of the SNP(s) (e.g., Ia4) (15).
Strain Clustering and SNP Analyses
Strain clustering and SNP analyses were performed as described (11). Briefly, clusters of related and singleton STs as well as evolutionary patterns among the isolates and for the entire dataset were determined by using eBURST (http://eburst.mlst.net). Neighbor-joining and minimum evolution methods in MEGA4 (http://www.megasoftware.net) were used to construct the trees along with multiple substitution models, including p-distance and Jukes-Cantor; all methods gave similar results. To test support for each node in the tree, we performed 1,000 bootstrap replicates.
All SNPs were identified for each ST by using the PROC FREQ tool in SAS software (SAS Institute, Cary, NC, USA). The probability of association of a SNP with an ST was determined by using a classification index (16). Variance across the dataset was determined using the Levene test (17). A p value <0.05 was considered significant.
MLST Discrimination of C. trachomatis
The Table shows the distribution of MLST STs and ompA genotypes for each dyad with SNP location(s), if present, for each of the 7 MLST housekeeping genes. We noted that in some cases, the DNA extracted directly from the patient sample was sufficient for MLST, whereas in other cases, the cultured sample was required because the patient sample did not yield sufficient DNA for MLST. We found no differences in MLST results when we compared DNA directly extracted from the patient sample with DNA extracted from the culture of the same sample.
For each of the 28 dyads, both partners had the same MLST ST. A total of 9 STs were found in the 56 samples from the 28 dyads: ST15, ST19, ST23, ST34, ST39, ST45, ST46, ST47, and ST55. Twenty-one dyads harbored MLST STs that matched STs of samples obtained from geographically diverse areas currently included in the MLST database, whereas 7 dyads harbored newly identified STs not present in the database (Technical Appendix Table 2). Among these 7 dyads, 4 MLST STs were unique to Indianapolis: ST45 (dyad 22), ST46 (dyads 23–26), ST47 (dyad 27), and ST55 (dyad 28); these results reflect the SNPs in various alleles (Table).
Eleven ompA genotypes were represented in the study and, by study design, were identical within dyads. The ompA genotypes included E, F, H, Ia, J, and K sequences that were identical to those of reference strains and 5 variant ompA genotypes of D1, D2, E6, F4, and Ia4 that had SNPs compared with the reference strains.
By combining MLST ST and ompA genotype data, we identified 13 unique strains (9 by MLST and 4 by ompA genotype) among the samples from our study group. Among these 13 strains, 8 were unique to Indianapolis and were found in 12 dyads (dyads 1, 6, 9, 12, 18, 22, 23, 24, 25, 26, 27, and 28) (Table; Technical Appendix Table 2). Moreover, of the 13 strains identified among the dyad samples, 9 (69%) contained gene sequences that suggested recombination within the genome, meaning that the ompA genotype was different from the ompA genotype that should be associated with the MLST ST if the genome were just 1 strain. For example, for strains from dyads 10 and 12, the ompA was D2, but the sequences of the 7 housekeeping genes (MLST ST34) matched the 7 housekeeping genes of strain F/ICCal3 from the MLST database. Putative recombinants (boldface in Table) represented a rate of 32% (9 of 28 samples).
MLST STs and ompA genotypes for each sample in the MLST dataset are shown in Technical Appendix Table 2. We found substantial variability of ompA genotypes associated with MLST STs, as shown previously (11). For the STs for the 56 Indianapolis samples, ST15 was associated with ompA genotypes J and K; ST34 was associated with ompA genotypes D2, E, and F; and ST19 was associated with ompA genotypes D, G, H, I, and J. online Technical Appendix Table 3 shows the characteristics of the alleles for each MLST locus based on the inclusion of the Indianapolis dataset in the MLST database.
Phylogeny of STs by Disease Phenotype and Evidence for Recombination
Figure 1

Figure 1. Population snapshot for Chlamydia trachomatis samples collected during April 2000–October 2003 from members of heterosexual partnerships (dyads) in Indianapolis, Indiana, USA, compared with reference strains. Data were compiled in eBURST (<>
The association of disease phenotype with 3 clonal complexes (CCs) was identified by eBURST (Figure 1), similar to those we reported previously: C. trachomatis strains that cause trachoma A, B, Ba, and C (CC-A); noninvasive STIs with low population prevalence (CC-B); and noninvasive, globally prevalent D/Da, E, and F STIs (CC-C). The Indianapolis strains were confined to noninvasive STI CCs (B and C), as expected. The strains associated with 3 of the 4 unique STs in the Indianapolis samples are seen in CC-C (Figure 1).
Figure 2
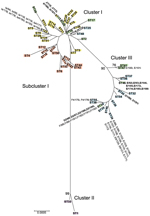
Figure 2. Minimum evolution tree of Chlamydia trachomatis samples collected during April 2000–October 2003 from members of heterosexual partnerships (dyads) in Indianapolis, Indiana, USA, compared with reference strains. The tree was constructed by...
The minimum-evolution tree also displayed 3 disease clusters (Figure 2); each of the Indianapolis strains is denoted next to the corresponding ST. Cluster I grouped noninvasive, low-prevalence STIs (eBURST CC-B), including a subcluster of strains that cause trachoma (eBURST CC-A). Cluster II grouped only invasive LGV strains. Cluster III grouped noninvasive, prevalent D/Da, E, and F STIs (eBURST CC-C). The tree constructed based on amino acid analysis showed similar clustering (data not shown).
The 9 putative Indianapolis recombinants were localized on the MLST tree with strains of the same ompA genotype and were recombinants of strains within the same cluster. Most recombinants were in cluster III. Four dyads with ST46 (unique to Indianapolis) had ompA genotype E and homology of the 7 housekeeping genes to reference strain Da/TW448 but with SNPs in leuS. D1/90i and D1/91i (ST45) and D2/96i, D2/97i, D2/189i, and D2/190i (ST34) were recombinants with homology to F/ICCal3 with SNPs in glyA and F/ICCal3, respectively. In cluster I, J/112i and J/113i shared the same ST as K/186i and K/187i and were recombinants with K/42nl and K/49nl. H/114i and H/115i were recombinants with G/SotonG1.
We investigated a well-defined, epidemiologically linked partner cohort of persons with C. trachomatis infection in which members of each dyad shared strains with identical ompA genotype and found that MLST ST was identical as well. This study confirms the reproducibility of MLST and short-term (≈30 days) stability of MLST in the context of a sexual partnership in which transmission has likely occurred. Whereas the identification of 8 unique C. trachomatis strains in Indianapolis was not surprising, given that samples from this city had not previously been subjected to MLST, the rate of 32% (9/28 samples) for recombinants was striking. We only considered 28 dyads in our analyses of strain diversity because of the closely defined epidemiologic link within the partnerships and the fact that strains were identical within dyads.
Becuase MLST provides >3 times the genetic data of ompA, the additional discriminatory power of this typing method is not surprising. We found that 8 of the 28 Indianapolis dyads (dyads 1 and 22–28) contained ompA genotypes that did not match our previous associations of ompA genotype with MLST STs in the MLST database (Table). For example, a J strain by ompA genotyping was associated with the MLST ST of a K strain (dyad 1); an ompA E strain was associated with the MLST ST of a Da strain (dyads 23–26); an ompA D1 strain was associated with the MLST ST of an F strain with SNPs in glyA (dyad 22); an ompA E strain was associated with the MLST ST of an E strain with 3 SNPs in hybG and 2 in pykF (dyad 27); and an ompA strain F4 matched the MLST of an F strain with an SNP in leuS (dyad 28).
ompA genotyping should not be considered a formal part of an MLST scheme because it is under immune selection (18), and housekeeping genes provide a stable evolutionary marker for STs. However, ompA genotyping remains a useful tool because it has been the mainstay of typing C. trachomatis for >20 years and is valuable for comparison with strains typed only by this method. Furthermore, ompA genotyping, but not MLST, was able to identify 2 dyads in which partners were infected with strains exhibiting mutations in ompA that had not previously been detected: E6 (dyad 18) and Ia4 (dyad 6) (Table). This result indicates utility in continuing ompA genotyping as a separate but adjunctive method with MLST for epidemiologic and transmission studies and for establishing strain concordance among members of less well-defined partnerships.
We further identified 3 clonal complexes that correlated with phenotypic disease, similar to previous findings (11). Most Indianapolis samples clustered with noninvasive D/Da, E, and F strains in CC-C (Figure 1), a result that is expected, given that these strains are the most prevalent worldwide (19–22). Whereas the Indianapolis samples were represented in 9 STs, 4 of these STs were distinct for this city, which suggests some clonal expansion of those unique strains in this area.
Genomic characteristics may also drive specific events, such as recombination, that may result in clonal expansion within a relatively small sexual network. Recombinants of the most prevalent urogenital ompA genotypes E, F, and D have previously been reported (23,24). Several reports have also been published regarding recombinants between genotypes D, E, and F and ompA genotype J (11,24,25); recombinants of LGV and D strains have also been documented (6,10,23), and previous MLST studies have shown evidence for recombination (7,10,11). In a previous study, we found 9 (17%) of 53 urogenital samples, excluding all ocular samples from patients with trachoma, were recombinant among a geographic distribution that included the western United States, Portugal, the Netherlands, and Ecuador (11).
In this study, members of 9 (32%) of the 28 dyads were infected with strains that contained gene sequences suggesting recombination within the genome (Table); this was the case for 2 of 4 STs that were unique to Indianapolis (STs 45 and 46). The 9 putative recombinants were localized on the MLST tree with strains of the same ompA genotype and, not surprisingly, were recombinants of strains within the same cluster (Figure 1). Our sample size was small, but the high rate of recombination suggests emerging diversity within a tight sexual network. This hypothesis is supported by historic studies of ompA genotypes among patients attending inner city STD clinics; in one such example, Ia genotypes that have much lower prevalence in other parts of the United States predominated among patients in Birmingham, Alabama, and were more prevalent than genotype D, which was the third most prevalent genotype for all other cities studied (26).
Our study has several strengths. Availability of the concordance study with carefully defined sexual partnerships, application of MLST to confirm the concordance of samples between dyads and to identify unique strains, and use of full-length ompA sequences enabled us to identify ompA variants and compare and combine the results of the 2 strain typing methods. The weaknesses of our study include the relatively small numbers of sexual partners and that only concordant dyads with epidemiologically linked strains were studied, limiting our conclusions about the overall diversity of strains in Indianapolis, which are likely much larger than what we discovered here.
In summary, our findings validate the discriminatory power of MLST for partnership and transmission studies of C. trachomatis infections among at-risk populations locally and globally. Larger partner and population studies that use MLST and ompA genotyping will provide valuable data on transmission concordance or discordance that will inform interventions and public health policy to better control C. trachomatis transmission. Furthermore, applying these tools globally will expand our knowledge of C. trachomatis strain diversity and their emergence among populations at risk for chlamydial STIs.
Dr Batteiger is an academic infectious diseases physician in the Division of Infectious Diseases, Department of Medicine, and Department of Microbiology and Immunology at Indiana University School of Medicine. His research interests are the epidemiology and molecular epidemiology of C. trachomatis and other sexually transmitted infections in the context of longitudinal studies of high-risk adolescents and infection concordance in partnerships.
Acknowledgments
We thank James Rothschild for excellent technical support.
This work was supported in part by Public Health Service grants from the National Institutes of Health (R01 AI098843 to D.D. and U19 AI31494 to J.D.F. and B.E.B.) and a grant from the Centers for Disease Control and Prevention (UR3/CCU5516481 to B.E.B.).
References
- World Health Organization. Global incidence and prevalence of selected curable sexually transmitted infections—2008. World Health Organization, Department of Reproductive Health and Research; 2012 [cited 2014 Mar 30]. http://www.who.int/reproductivehealth/publications/rtis/stisestimates/en/index.html
- Satterwhite CL, Torrone E, Meites E, Dunne EF, Mahajan R, Ocfemia MC, Sexually transmitted infections among US women and men: prevalence and incidence estimates—2008. Sex Transm Dis. 2013;40:187–93. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Division of STD Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention. Sexually transmitted disease surveillance 2011. Atlanta (GA): US Department of Health and Human Services; 2012.
- Gottlieb SL, Martin DH, Xu F, Byrne GI, Brunham RC. Summary: the natural history and immunobiology of Chlamydia trachomatis genital infection and implications for Chlamydia control. J Infect Dis. 2010;201(Suppl 2):S190–204. DOIPubMedGoogle Scholar
- Klint M, Fuxelius HH, Goldkuhl RR, Skarin H, Rutemark C, Andersson SG, High-resolution genotyping of Chlamydia trachomatis strains by multilocus sequence analysis. J Clin Microbiol. 2007;45:1410–4. DOIPubMedGoogle Scholar
- Somboonna N, Wan R, Ojcius DM, Pettengill MA, Joseph SJ, Chang A, Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L(2)) and D lineages. MBio. 2011;2:e00045–11. DOIPubMedGoogle Scholar
- Pannekoek Y, Morelli G, Kusecek B, Morre SA, Ossewaarde JM, Langerak AA, Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 2008;8:42. DOIPubMedGoogle Scholar
- Christerson L, Bom RJ, Bruisten SM, Yass R, Hardick J, Bratt G, Chlamydia trachomatis strains show specific clustering for men who have sex with men compared to heterosexual populations in Sweden, the Netherlands, and the United States. J Clin Microbiol. 2012;50:3548–55. DOIPubMedGoogle Scholar
- Gravningen K, Christerson L, Furberg AS, Simonsen GS, Odman K, Stahlsten A, Multilocus sequence typing of genital Chlamydia trachomatis in Norway reveals multiple new sequence types and a large genetic diversity. PLoS ONE. 2012;7:e34452. DOIPubMedGoogle Scholar
- Bom RJ, van der Helm JJ, Schim van der Loeff MF, van Rooijen MS, Heijman T, Matser A, Distinct transmission networks of Chlamydia trachomatis in men who have sex with men and heterosexual adults in Amsterdam, The Netherlands. PLoS ONE. 2013;8:e53869. DOIPubMedGoogle Scholar
- Dean D, Bruno WJ, Wan R, Gomes JP, Devignot S, Mehari T, Predicting phenotype and emerging strains among Chlamydia trachomatis infections. Emerg Infect Dis. 2009;15:1385–94. DOIPubMedGoogle Scholar
- Khan A, Fortenberry JD, Juliar BE, Tu W, Orr DP, Batteiger BE. The prevalence of chlamydia, gonorrhea, and trichomonas in sexual partnerships: implications for partner notification and treatment. Sex Transm Dis. 2005;32:260–4. DOIPubMedGoogle Scholar
- Stothard DR, Boguslawski G, Jones RB. Phylogenetic analysis of the Chlamydia trachomatis major outer membrane protein and examination of potential pathogenic determinants. Infect Immun. 1998;66:3618–25 .PubMedGoogle Scholar
- Batteiger BE, Tu W, Ofner S, Van Der Pol B, Stothard DR, Orr DP, Repeated Chlamydia trachomatis genital infections in adolescent women. J Infect Dis. 2010;201:42–51. DOIPubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Jolley KA, Wilson DJ, Kriz P, McVean G, Maiden MC. The influence of mutation, recombination, population history, and selection on patterns of genetic diversity in Neisseria meningitidis. Mol Biol Evol. 2005;22:562–9. DOIPubMedGoogle Scholar
- Levene H. Robust Tests for Equality of Variances. In: Olkin I, editor. Contributions to probability and statistics: essays in honor of Harold Hotelling. Stanford (CA): Stanford University Press; 1960. p. 278–92.
- Ward ME, Treharne JD, Murray A. Antibody specificity of human antibody to Chlamydia in trachoma and lymphogranuloma. J Gen Microbiol. 1986;132:1599–610 .PubMedGoogle Scholar
- Korhonen S, Hiltunen-Back E, Puolakkainen M. Genotyping of Chlamydia trachomatis in rectal and pharyngeal specimens: identification of LGV genotypes in Finland. Sex Transm Infect. 2012;88:465–9. DOIPubMedGoogle Scholar
- Fieser N, Simnacher U, Tausch Y, Werner-Belak S, Ladenburger-Strauss S, von Baum H, Chlamydia trachomatis prevalence, genotype distribution and identification of the new Swedish variant in Southern Germany. Infection. 2013;41:159–66. DOIPubMedGoogle Scholar
- Machado AC, Bandea CI, Alves MF, Joseph K, Igietseme J, Miranda AE, Distribution of Chlamydia trachomatis genovars among youths and adults in Brazil. J Med Microbiol. 2011;60:472–6. DOIPubMedGoogle Scholar
- Wang Y, Skilton RJ, Cutcliffe LT, Andrews E, Clarke IN, Marsh P. Evaluation of a high resolution genotyping method for Chlamydia trachomatis using routine clinical samples. PLoS ONE. 2011;6:e16971. DOIPubMedGoogle Scholar
- Millman KL, Tavare S, Dean D. Recombination in the ompA gene but not the omcB gene of Chlamydia contributes to serovar-specific differences in tissue tropism, immune surveillance, and persistence of the organism. J Bacteriol. 2001;183:5997–6008. DOIPubMedGoogle Scholar
- Jeffrey BM, Suchland RJ, Quinn KL, Davidson JR, Stamm WE, Rockey DD. Genome sequencing of recent clinical Chlamydia trachomatis strains identifies loci associated with tissue tropism and regions of apparent recombination. Infect Immun. 2010;78:2544–53. DOIPubMedGoogle Scholar
- Gomes JP, Bruno WJ, Nunes A, Santos N, Florindo C, Borrego MJ, Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 2007;17:50–60. DOIPubMedGoogle Scholar
- Millman K, Black CM, Johnson RE, Stamm WE, Jones RB, Hook EW, Population-based genetic and evolutionary analysis of Chlamydia trachomatis urogenital strain variation in the United States. J Bacteriol. 2004;186:2457–65. DOIPubMedGoogle Scholar
Figures
Table
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 20, Number 11—November 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Deborah Dean, Children’s Hospital Oakland Research Institute, 5700 Martin Luther King Jr Way, Oakland, CA 94609, USA
Top